
Título
Autor
Lugar de Realización
Correspondencia
Texto
COMPRESIONES NERVIOSAS PERIFERICAS MÚLTIPLES DE ORIGEN METABÓLICO: MUCOPOLISACARIDOSIS. DESCRIPCIÓN DE UN CASO Y REVISIÓN DE LA LITERATURA
Gabriel Moreno1, Silvina Sánchez2, Mariano Socolovsky1, Hernán Gonza1, Mirta Amores2, Javier Gardella1
1: Servicio de Neurocirugía. Instituto de Neurociencias, 2: Servicio de Neurología, Instituto de Neurociencia, Hospital de Alta Complejidad "Juan Domingo Perón", Formosa, Argentina.
RESUMEN
Introducción. Las enfermedades por depósito de Glucosaminoglicanos en el tejido conectivo se asocian a neuropatías compresivas diversas, especialmente al síndrome del túnel carpiano y del túnel tarsiano. La afectación nerviosa es precoz dentro de la evolución de la enfermedad y suele dejar secuelas permanentes.
Descripción. Se presenta el caso de una joven de 20 años con Mucopolisacaridosis tipo VI (síndrome de Maroteaux-Lamy), en terapia de reemplazo enzimático de 3 años de evolución, que presentó síndrome del túnel carpiano bilateral y del túnel tarsiano derecho.
Intervención. El diagnóstico se confirmó neurofísiológicamente y la descompresión quirúrgica brindó resultados clínicos muy favorables, mejorando notablemente la calidad de vida de la enferma.
Conclusión. Esta rara patología y su asociación con las neuropatías compresivas debe ser conocida por neurocirujanos, ya que suele requerir tratamiento quirúrgico por la especialidad.
Palabras clave: Compresión nerviosa crónica, Mucopolisacaridosis, Enfermedad de Hurler
Correspondencia:
socolovsky@fibertel.com.ar
Recibido: agosto de 2011.
Aceptado: septiembre de 2011.
INTRODUCCIÓN
Las compresiones nerviosas crónicas son lesiones adquiridas en la edad adulta, siendo muy frecuentes a partir de la cuarta y quinta décadas de la vida. Muy raramente se presentan en la población pediátrica o adolescente. Cuando lo hacen, muchas veces se encuentran asociadas a diversas enfermedades metabólicas; congénitas. Este trabajo presenta un caso de una paciente de 17 años de edad que sufría de mucopolisacaridosis (MPS) tipo VI, conocida como enfermedad de Maroteaux- Lamy, causada por deficiencia de la enzima Arilsulfatasa B y que suele producir trastornos óseos, baja talla y opacidad corneal con déficit visual, no estando asociada. a alteraciones del desarrollo intelectual1-4. Como complicación de esta enfermedad. la paciente presentó síndrome del túnel carpiano bilateral y síndrome del túnel del tarso, también bilateral, los cuales fueron tratados quirúrgicamente. El objetivo de esta presentación es llamar la atención a los neurocirujanos respecto de esta patología, así como la necesidad, en muchos casos, de descomprimir los nervios periféricos si se encuentran afectados, para evitar déficits funcionales permanentes. Dado que el interrogatorio es dificil en estos pacientes, y que raramente refieren en forma espontánea síntomas que eleven la sospecha clínica, es importante que quienes traten esta patología tengan en cuenta la frecuente asociación de la enfermedad con síndromes compresivos de los nervios periféricos, como paso inicial para un buen diagnóstico y tratamiento.
DESCRIPCIÓN
Paciente de 20 años con Síndrome de Maroteaux-Lamy. Presenta facies característica de esta enfermedad (Fig. 1), alteraciones generalizadas del desarrollo óseo (Fig. 2), siendo su altura de 1,02 m. Se encuentra en tratamiento de reemplazo enzimático desde hace tres años, lo cual mejoró sus condiciones físicas generales as.i como el nivel de GAGs urinarios excretados. Pese a esta evolución favorable, presenta en forma asociada síndrome del túnel carpiano bilateral con diagnóstico neurofisiológico por disminución de la velocidad de conducción y alteraciones electromiográficas. La clínica es atípic, por presentar escaso dolor, signo de Tinel y leves parestesias, que se encuentran enmascarados por la displasia ósea, la rigidez articular y la cronicidad de los síntomas, lo cual dificulta su manifestación por parte del paciente. Durante el tratamiento de reemplazo enzimático presentó dolor en planta de pie derecho e imposibilidad de deambular, con exacerbación del dolor a la percusión en la región del nervio tibial posterior. Se confirmó neurofisiológicamente síndrome del túnel tarsiano. Se realizó cirugía descompresiva en dos tiempos: en el primero se realizó descompresión del nervio mediano izquierdo, y en el segundo neurolisis del nervio mediano (Fig. 3) y el nervio tibial posterior derechos (Fig. 4). En la evolución inmediata de la liberación del primer túnel carpiano - izquierdo- la paciente refiere desaparición de parestesias y mejoría en la sensibilidad de la mano y antebrazo, déficit del cual la paciente refiere no haber sido consciente: desde el punto de vista motor, la paciente refiere mejoría de la prensión. El control neurofisiológico, 3 meses después de la cirugía, no evidenció mejoría. Luego de la liberación del túnel carpiano derecho la paciente no refirió una mejoría tan clara como cuando fuera liberado el lado izquierdo, sin embargo la liberación del nervio tarsiano tuvo una recuperación inmediata del dolor, lo que posibilitó la deambulación nuevamente.

Fig. 1 . Facies característica del síndrome de Maroteawc-Lamy que presenta la paciente reportada en este trabajo.

Fig. 2. Trastornos del desarrollo óseos característicos: ambas manos de la paciente en estudio, vista dorsal.
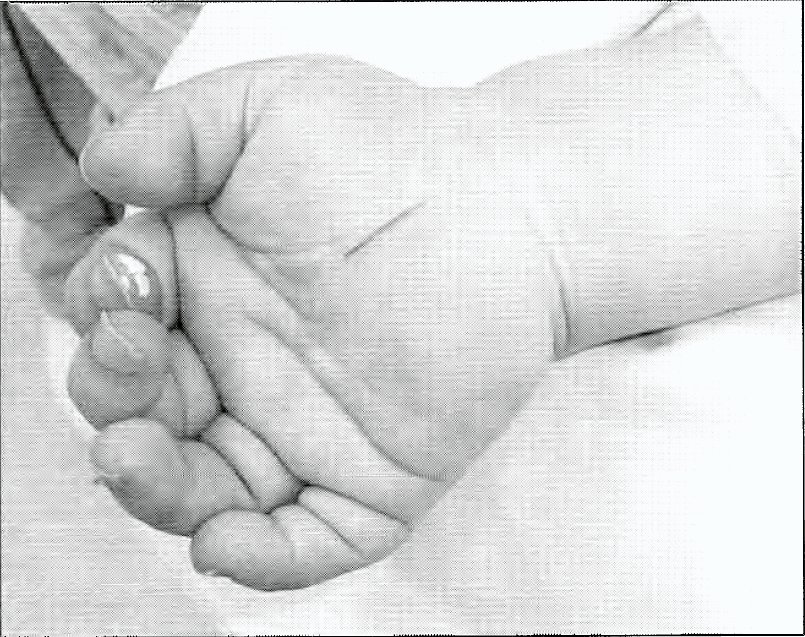
Figura 3: Mano derecha en posición quirúrgica con la incisión para descomprimir el nervio mediano ya marcada
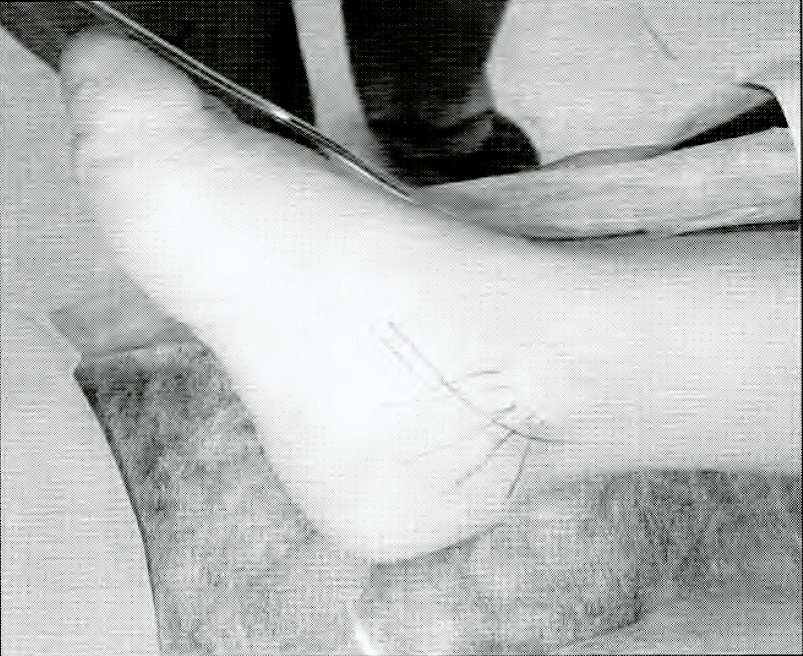
Fig. 4. Cara interna del pie derecho, con incisión marcada para descomprimir el nervio tibial posterior.
DISCUSIÓN
Diagnóstico y clínica de los síndromes compresivos en las MPS.
Las MPS son un grupo de enfermedades metabólicas infrecuentes que comparten un hecho en común: tienen una deficiencia congénita de una o más enzimas que efectúan el ciclo de los Glicosaminoglicanos (GAG) lisosomal. Como consecuencia de ello, se acumulan GAGs en estado natural en el tejido conectivo en forma progresiva e ininterrumpida, lo cual provoca innumerables trastornos en órganos y tejidos. Los pacientes con MPS con diferentes grados de severidad comparten ciertas características clínicas: fascies típica, discapacidad visual, hepatoesplenomegalia, rigidez articular, dismorfias esqueléticas, y patología cardiorrespiratoria. Asimismo, la mayoría de las MPS se asocian con trastornos mentales y retraso, salvo la MPS tipo IV Ay B y la MPS VI y las formas leves de la MPS I y II. Hay un amplio espectro de las tasas de progresión dentro de cada tipo de MPS1-4.
Las MPS son una causa común de STC, síndrome del túnel tarsiano y otras neuropatías compresivas crónicas en niños y adolescentes5-13. Se han diagnosticado compresiones asociadas a esta enfermedad tan tempranamente como a los dos años de vida. Como las MPS son de carácter progresivo, es esencial que los síndromes compresivos sean diagnosticados y tratados antes de que los daños en el nervio afectado se conviertan en irreversibles. Sin embargo, en la actualidad no existen normas para el diagnóstico y el tratamiento del STC en la MPS. El diagnóstico correcto del STC en general implica la evaluación de los signos y síntomas en combinación con los estudios de conducción del nervio. Particularmente en el caso de la MPS, los signos y síntomas del STC están ausentes a diferencia que en el paciente adulto. El diagnóstico precoz en estos niños a menudo se retrasa porque los signos típicos como dolor nocturno, entumecimiento, hormigueo, signo de Tinel, prueba de Phalen, etc, tienden a estar ausentes11,12. El reconocimiento de síntomas sutiles como la disminución de la sudoración, despertares nocturnos, mordeduras de las manos y torpeza manual adquieren una mayor importancia. Es probable que los signos clásicos del STC, por dar un ejemplo, sean enmascarados por otros hallazgos de MPS como la rigidez articular y la displasia esquelética. Además, el inicio es más gradual, lo que puede contribuir a la falta de quejas subjetivas y el reconocimiento de los signos y síntomas clínicos puede ser confundido por la incapacidad de los niños a comunicar sus problemas, lo que ciertamente se aplica a niños muy pequeños o con retraso en el desarrollo. Debido a ello, es frecuente que las neuropatías compresivas no sean reconocidas hasta que esté presente la hipotrofia o la perdida de funciones sensitivas y/o motoras.
Diagnóstico neurofisiológico
El diagnostico precoz de las neuropatías compresivas es imperativo en los pacientes con MPS, dado que estos pacientes tienden a tener una buena respuesta funcional y recuperación neurofisiológica después de la cirugía 5,10,11. Existen varias pruebas para diagnosticar STC aunque ninguna de ellas lo hace en forma precoz. Sin embargo, los procedimientos habitualmente utilizados para el diagnóstico de STC en adultos no siempre son adecuados para los niños. A pesar de que actualmente no hay consenso sobre el método de elección para evaluar STC en los pacientes con MPS, la neurofisiología es lo más objetivo de que se dispone en la actualidad.
Las pruebas diagnósticas que se utilizan para evaluar la función periférica del nervio incluyen el estudio de la conducción nerviosa y la electromiografía. El gold standard para el diagnóstico de neuropatías compresivas en pacientes con MPS es el estudio de la velocidad de conducción. Se recomienda realizar dicho estudio como screennig de rutina en todos los pacientes con MPS cada uno a tres años, más allá de la evaluación clínica periódica, dada la incapacidad de estos pacientes para comunicar sus síntomas con claridad, ya sea por el retraso mental cuando está presente, o debido a otras causas más diversas (por ejemplo la precocidad y cronicidad de sus síndromes neurológicos compresivos. En muchas ocasiones los estudios resultan difíciles de realizar, ya sea por la falta de colaboración de los pacientes, las manos y pies pequeños, o la intolerancia a los pinchazos. En el caso de la electromiografía, suele requerirse sedación para realizarla.
TRATAMIENTO
Las técnicas no quirúrgicas para el tratamiento de las neuropatías compresivas, que incluyen reposo, férulas y AINE, pueden ser considerados sólo para los casos leves6. La cirugía está indicada en casi todos los casos con evidencia clínica y neurofisiológica de neuropatía compresiva y consiste en una neurolisis. Históricamente, la descompresión quirúrgica ha sido y es indicada en forma precoz una vez que se arriba al diagnóstico, por dos razones: 1) las posibilidades de que el daño al nervio sea irreversible en ese momento no son bajas2,9, y 2) el tratarse de una enfermedad que lejos de estabilizarse, continúa su progresión a medida que los depósitos de GAG en el tejido conectivo continúan incrementándose.
Diversos estudios han demostrado mejoría clínica dé la función de la mano luego de la liberación del nervio mediano en el túnel carpiano en niños con MPS11 sin estar acompañada de mejoría neurofisiológica8,9, como es el caso presentado en este trabajo. Haddad14 reportan los resultados de la descompresión temprana en 48 niños con STC asociado a MPS o ML, demostrando que los casos leves con STC tenían una mejor recuperación neurofisiológica que los casos severos. En un estudio, sólo 2 de 16 pacientes mostraron una mejoría electro fisiológica después de la descompresión del nervio mediano, mientras que la mayoría de los pacientes y sus padres reportaron mejoría de los síntomas13. Por el contrario, todos los pacientes mostraron normalizados o mejorados los resultados electro fisiológicos después de la cirugía en un estudio realizado por Khanna15.
El advenimiento del transplante de células madres hematopoyéticas (HSCT) y la terapia de reemplazo enzimático para el tratamiento de los diferentes tipos de MPS ha cambiado considerablemente el pronóstico de calidad y cantidad de vida de los pacientes con MPS, pero hasta ahora ha fallado para prevenir el desarrollo del STC. Por ejemplo, en el estudio de Guffon et al16, se evaluaron los efectos del HSCT en 9 pacientes con síndrome de Hurler de los cuales 8 pacientes desarrollaron STC durante el curso de su enfermedad y todos ellos requirieron una liberación del mediano incluyendo a 5 después del inicio de HSCT. Del mismo modo, en el estudio de Khanna9, 12 de 17 pacientes con MPS que habían recibido previamente HSCT desarrollaron STC. Por el contrario, el transplante de 43 niños con síndrome de Hurler antes de los 2 años redujo el riesgo de desarrollo de STC en un 46%. El riesgo disminuyo en un 78% en los niños que tenían una actividad enzimática normal después de la HSCT. A pesar de esta evolución positiva los niños permanecieron en riesgo de desarrollar STC y los autores recomiendan monitoreo regular con estudios de conducción del nervio.
Poca información existe en la actualidad sobre el impacto de la terapia de reemplazo enzimático en el síndrome del túnel carpiano en los niños con MPS. Dado que esta terapia se aplica cada vez más en pacientes con MPS, existe la necesidad de investigación clínica en este tema. Si los resultados de la terapia mejoran la expectativa de vida, proponer tratamientos agresivos para las neuropatías compresivas será cada vez más importante para mantener la función de la mano durante el mayor tiempo posible.
CONCLUSIÓN
El caso presentado y el análisis de la literatura realizado en esta presentación, permiten establecer que el tratamiento de elección en la patología compresiva de los nervios periféricos asociada a enfermedades metabólicas como las Mucopolisacaridosis es quirúrgico. La cirugía debe ser realizada en forma precoz para evitar secuelas definitivas. El diagnóstico es dificil en los casos con mucha afectación clínica, por lo cual el médico tratante debe estar al tanto de esta información. El aumento de la expectativa de vida asociado a las nuevas terapias como el reemplazo enzimático obligan a ofrecer a estos pacientes opciones terapéuticas apropiadas al momento actual.
Agradecimiento
Los autores desean agradecer al Sr. Hugo Dasso y a la Lic. Mariela Handverker el apoyo brindado para la correcta atención del caso presentado.
Bibliografía
- Cathey S: Molecular order in mucolipidosis II and III nomenclature. Am.J. Med. Genet. 146A: 512-513, 2008.
- McKusick VA: Heritable Disorders of Connective Tissue. Ed. 4, pp 521-686. St. Louis, C. V. Mosby, 1972.
- Pastores, GM, KolodnyE: Lysosomal storage diseases. In PediatrIc Neurology: Principies & Practice. 4th Edition. K.F. Swaiman. S. Ashwal D.M. Ferriero, Eds.: Vol. 1: 659-714. Mosby Elsevier Philadelphia, 2006.
- Tylki-Szymanska, A: Clinical variability in mucolipidosis III (Pseudo- Hurler polydystrophy). Am J Med Genet 08 : 214-218, 2002.
- Albrektsson, B, Rydholm U: The tarsal tunnel syndrome in children. J. Bone Joint Surg. 64B:215-217, 1982.
- Barfred T, Ipsen T: Congenital Carpal Tunnel Syndrome. J. Hand Surg 10: 246-248, 1985.
- Danta G: Familial Carpa' Tunnel Syndrome with Onset in Childhood. J Neurol Neurosurg Psychiat 38: 350-355, 1975.
- Feingold MH, Hidvegi E, Horwitz SJ: Bilateral Carpal Tunnel Syndrome in an Adolescent. Am J Dis Child, 134: 394-396, 1980.
- Gschwind, C. Tonkin MA: Carpal tunnel syndrome in children with mucopolysaccharidosis and related disorders. J. Hand Surg. 17A: 44-47, 1992.
- Lettin AW: Carpal Tunnel Syndrome in Childhood. Report of a Case. J Bone and Joint Surg 47-B(3): 556-559, 1965.
- McArthur RG, Hayles AB, Gómez MR, Blanco AJ: Carpal Tunnel Syndrome and Trigger Finger in Childhood. Am J Dis Child, 117: 463-469, 1969.
- Miner ME, Schimke RN: Carpal Tunnel Syndrome in Pediatric Mucopolysaccharidoses. Report of Four Cases. J Neurosurg, 43: 102-103, 1975.
- Van Meir, NL, De Smet M: Carpal tunnel syndrome in children. J. Pediatr.Orthop. B. 14 : 42-45, 2005.
- Haddad, FS: Carpal tunnel syndrome in the mucopolysaccharidoses and mucolipidoses. J. Bone Joint Surg. 79B : 576-582, 1997.
- Khanna G, Van Heest AE, Angel J, Bjorarker K, Grewal S: Analysis of factors affecting development of carpal tunnel syndrome in patients with Hurler syndrome after hematopoietic cel transplantation. Bone Marrow Transplantation 2007; 39(6):331- 4
- GuffonN, Souillet G, Maire I, Straczek J, Guibaud P: Follow-up of nine patients with Hurler syndrome after bone marrow transplan
Abstract
Introduction. Diseases characterized by glycosaminoglucan stores in the connective tíssue are associated to compression neuropathíes and in particular to the carpal tunnel and the tarsal tunnel syndromes. Nerves are affected early in the course of the disease and regularly result in permanent sequelae.
Description. A case is presented of a young 20 year old female with mucopolysaccharidosis type TV (Maroteaux-Lamy syndrome) dating back 3 years treated with enzyme replacement therapy that presented with bilateral carpal tunnel and right tarsal tunnel syndrome.
Intervention. The diagnosis was confirmed through neurophysiological testíng and surgical decompression attained very favorable clinical results that dramatically improved her qualíty of life.
Conclusion. This rare disease and its association with compressive neuropathies should be well understood by neurosurgeons since it normally requires neurosurgery.
Key words: chronic nervous compression, mucopolysaccharidosis, Hurler's disease
COMENTARIO
Los doctores Moreno y colaboradores presentan el interesante caso de una joven paciente con una mucopolisacaridosis tipo VI o síndrome de Maroteaux Lamy asociado a neuropatías compresivas. Señalan asimismo que dado las dificultades en el interrogatorio o examen neurológico que pueden ofrecer estos pacientes, es necesario tener un alto grado de sospecha ante la frecuencia de estos problemas en las MPS y mucolipidosis (ML).
Los síndromes del tune del carpo (STC) raramente se presentan en la infancia y en una búsqueda bibliografiíta solo se encontraron algo mas de 200 casos publicados y la mayoría estaba asociados a un trastorno genético. Entre estos la asociación mas frecuente era con las enfermedades lisosomales de deposito como la MPS y las ML. Para complicar las cosas, como en este caso, la mayoría de los afectados no presenta los signos y síntomas típicos del STC del adulto sino que por el contrario solo se manifiesta por cierta torpeza motora para las tareas finas. Mucho mas complicado resulta la exporacion de este tipo de signos en el síndrome del túnel del tarso. Ante estos problemas, los autores sugieren efectuar en forma rutinaria y periódica, en esta población de alto riesgo, estudios neurofisiológicos para descartar esta patología. Sin duda la ventaja de esta metodología incluye el diagnostico precoz que conlleva mejores perspectivas de recuperación.
Vale la pena mencionar que si bien en la actualidad las perspectivas terapéuticas de estas enfermedades han variado, lamentablemente el transplante de medula ósea no parece prevenir alguna de estas neuropatías compresivas.
La publicación de este caso, sin duda sirve para alertar a neurólogos, clínicos y pediatras que tengan ocasión de tratar este tipo de pacientes, sobre la necesidad de estar advertidos sobre la alta posibilidad de que se presenten neuropatías compresivas y el valor que tiene su diagnostico y tratamiento precoz.
Federico Micheli