
Título
Autor
Fecha
Lugar de Realización
Texto
Artículo original
Tumores de la Región Pineal en Pediatría
J.C. Suárez, J.C. Viano, E.J. Herrera, S.B. Zunino
Unidad de Neurocirugía, Departamento de Cirugía, Hospital Infantil Municipal, Córdoba
RESUMEN
Se presentan 10 casos de tumores de la región pineal en pacientes con síndrome de hipertensión endocraneana y síndrome de Parinaud tratados entre abril de 1972 y marzo de 1994, con franco predominio (7:3) del sexo masculino.
El diagnóstico se realizó por TAC y IRM y en el 50% de los casos mediante marcadores tumorales (alfafetoproteínas).
Se analizan los resultados del tratamiento que combinó cirugía por distintas vías, radioterapia y quimioterapia.
No hubo mortalidad quirúrgica. Sobreviven 5 pacientes con una sobrevida promedio de 66 meses. La sobrevida promedio de los que fallecieron fue de 19 meses.
Palabras clave: región pineal, tumores, pineocitoma, germinoma, teratoma
ABSTRACT
The study covers 10 cases of tumors in the pineal area in patients with intracranial hypertension syndrome and Parinaud's syndrome treated between April 1972 and March 1994, with a significant majority of males (7:3).
The diagnosis was made using CAT and MRI and in 50% of the cases using tumoral markers (alphaphetoproteins).
An analysis is made of the results of the treatment, which combined surgery by different paths, radiotherapy and chemotherapy.
There was no surgical mortality. Five patients are still alive with an average survival of 66 months. The average survival of the decease patients was 19 month.
Key words: Pineal region, Germinoma, Neoplasms, Pineal Body, Teratomas.
INTRODUCCION
Los tumores originados en la región pineal son raros, representan el 0,4 al 4,5% de todos los tumores cerebrales11.
La incidencia de los tumores germinales en esta región es marcadamente mayor en las casuísticas japonesas (2,6 al 6,5%) que en las de los países occidentales (0,4 al 0,7%)2, 9, 38.
El 50% de estos tumores se diagnostican en pacientes menores de 20 años y en la población pediátrica representan entre el 3 y 8% de los tumores cerebrales14.
Las manifestaciones clínicas son tan variadas como el tipo de tumores que pueden desarrollarse en esta área. La hipertensión endocraneana es la más frecuente, pero también se presentan síntomas y signos provocados por infiltración o compresión de las estructuras vecinas, tales como: tálamo, óptico, cápsula interna, hipotálamo, fascículo talamomamilar, tubérculos mamilares, pedúnculos cerebrales y cerebelosos, etc.28.
El objetivo de esta publicación es presentar nuestra experiencia en el tratamiento de los tumores de esta región, en 10 pacientes tratados en la Unidad de Neurocirugía del Hospital Infantil Municipal de Córdoba, Argentina.
MATERIAL Y METODOS
Desde abril de 1972 a marzo de 1994 se internaron en la Unidad de Neurocirugía del Hospital Infantil Municipal de Córdoba 200 niños con tumores intercraneanos, de los cuales 10 tenían un tumor en la región pineal. Se revisaron retrospectivamente las historia clínicas de dichos pacientes, analizando sus síntomas, evolución y tratamiento.
El diagnóstico fue hecho con la clínica y los estudios complementarios: radiografías simples de cráneo en todos, ventriculografia en 2, tomografia axial computada (TAC) en 8, imágenes por resonancia magnética (IRM) en 4 y marcadores biológicos en 5 (alfafetoproteína y subunidad beta de la gonadotrofina coriónica).
En 9 pacientes se hizo cirugía del tumor con confirmación histológica. En 5 pacientes se realizó una derivación de líquido cefalorraquídeo (LCR): en uno como única cirugía, en 3 luego de una resección incompleta y en otro prequirúrgico (colocada en otra institución).
Todos los pacientes fueron irradiados con un equipo de CO 60, con una distancia frente-piel (DFP) de 80 cm y una dosis que varió según la histología del tumor y la edad del paciente. Se hicieron 35-40 Gy en encéfalo total, con 15-20 Gy de sobredosis tumoral y 35 Gy en médula espinal en los tumores de células germinales y en el ependimoma. En los demás la irradiación fue localizada en la región pineal con 50 Gy.
Se efectuó quimioterapia en 4 pacientes. El protocolo utilizado en el teratoma maligno fue: etoposido 100 mg/m2 los días P a 3° y cisplatino 90 mg/m2 el día 1° con 3 semanas de descanso, efectuando 9 ciclos. El protocolo utilizado en los pineoblastomas, en la década del 70, consistió en: CCNU 100 mg/m2/ler día, vincristina 1,5 mg/m2/días 1° al 45°, con 3 semanas de descanso, efectuando 6 a 9 ciclos.
RESULTADOS
Los tumores de la región pineal representaron el 5% de todos los tumores intracraneanos de nuestro servicio. Las edades oscilaron entre 2 y 15 años (media: 10 años), 7 fueron varones y 3 mujeres. Todos presentaron un síndrome de hipertensión endocraneana y sólo 4 un síndrome de Parinaud. Ninguno tuvo síntomas endocrinos (Tabla 1). El tiempo de evolución varió entre 7 días y 3 meses (media: 41,5 días).
Tabla 1. Tumores región pineal. Sintomatología

Las Rx simples de cráneo mostraron signos de hipertensión endocraneana en todos y calcificaciones de la región pineal en 2 (1 germinoma y 1 sin confirmación histológica). La TAC y la IRM mostraron, en todos los casos, el tumor asociado a hidrocefalia por obstrucción del acueducto de Silvio. Sólo en 1 caso hubo aumento de la alfafetoproteína (teratoma maligno); en los demás el dosaje de marcadores fue normal (germinoma, teratoma benigno, ependimoma y el caso sin confirmación histológica).
La resección quirúrgica fue total en 5 y subtotal en 5. El diagnóstico patológico fue: pineoblastomas 3, teratoma maligno (con áreas de germinoma) 1, astrocitoma grado 1 uno, astrocitoma anaplásico 1, ependimoma 1 y germinoma 1. No hubo mortalidad quirúrgica. Fallecieron 5 pacientes: 3 por metástasis craneoespinales (2 pineoblastomas y 1 terastoma maligno) y 2 por recidiva del tumor (1 pineoblastoma y el astrocitoma anaplásico).
Dos pacientes tuvieron depresión medular con la radio y quimioterapia. Uno, con un pineoblastoma, mejoró al suspender transitoriamente la radioterapia y otro, con el terastoma maligno, necesitó un trasplante de médula ósea.
La sobrevida de los pacientes fallecidos fue de 6 a 36 meses, con una media de 19 meses. Dos enfermos fallecieron antes del año de operados. Ambos tuvie- ron un pineoblastoma. Los 3 restantes fallecieron después del año de operados (pineoblastoma, terastoma maligno y astrocitoma anaplásico).
En los pacientes que viven, la sobrevida oscila entre 12 y 105 meses, con una media de 66 meses. De éstos, 3 tienen un examen neurológico normal y 2 presentan secuelas: 1 déficit intelectual y 1 trastornos de la deglución y fonación. Consideramos que estas secuelas son secundarias a la radioterapia.
DISCUS1ON
En nuestra serie el síndrome de hipertensión endocraneana se observó en el 100% de los casos. Fue producido por una hidrocefalia, secundaria a una obstrucción en la circulación del L.C.R. a nivel del acueducto de Silvio2, 5, 7, 9, 28, 38. Como signo focalizador observamos en 4 pacientes el síndrome de Parinaud5, 7, 28.
Los estudios neurorradiológicos modernos utilizados en esta muestra fueron la TAC de cerebro y la IRM que nos permitieron visualizar el tumor y la hidrocefalia concomitante. La IRM fue el procedimiento que más información nos brindó acerca de los elementos constitutivos del tumor, de su extensión y de la presencia o no de metástasis craneoespinales, obviando muchas veces la angiografía cerebral, como opinan varios autores5, 9, 11, 30, 39, 40, 41.
Los marcadores tumorales se dosaron en 5 pacientes de los cuales sólo en uno se detectó un nivel elevado de alfa fetoproteina37. Esto confirmaría la controversia sobre el valor de los marcados tumorales, que son de utilidad en el diagnóstico pre operatorio de los coriocarcinomas, carcinomas embrionarios y tumores del saco vitelino o tumores del seno endodérmico5, 7, 10, 14.
En nuestra serie los pineoblastomas representan el 30%; este tumor es incluido actualmente dentro de los tumores neuroectodérmicos primitivos, junto con el retinoblastoma, el neuroblastoma de cerebro, el meduloblastoma de cerebelo y el ependimoblastoma3, 25. No se asocian con elevación de los marcadores tumorales y no tienen preferencia de sexo; se los encuentra en pacientes jóvenes; son de evolución corta por su crecimiento rápido y por diseminarse a través del L.C.R. Histológicamente se caracterizan por la presencia de rosetas de Homer Wright y células gigantes7. El pronóstico es bueno en los teratomas benignos resecados totalmente; los resecados subtotalmente recidivan tardíamente25. Los teratomas inmaduros o malignos elevan los niveles de alfafetoproteínas, recidivan rápidamente y producen metástasis por el L.C.R. y fuera del sistema nervioso central4, 7. (Fig.1)
En nuestra experiencia aparecen los astrocitomas y ependimomas, cuyo manejo fue descripto en otras publicaciones31, 32. Al respecto deseamos enfatizar nuevamente la conveniencia de la resección total de los astrocitomas grado I y en los resecados subtotalmente la necesidad del control clínico y neurorradiológico periódico, para detectar precozmente su recidiva y reoperarlos con las técnicas neuroquirúrgicas modernas (microcirugía, coagulación bipolar y aspiración ultrasónica por cavitación o láser)15, 33, porque son tumores radiorresistentes y por los efectos secundarios de la radioterapia, observados en dos de nuestros pacientes1, 13, 21, 23, (Fig, 2)

Fig. 1: Paciente de 14 años, sexo masculino, con teratoma benigno de región pineal. A) RNM. B) Histología.
Los germinomas forman el 10% de nuestra muestra, pero su incidencia entre los tumores de la región pineal es del 50% en las publicaciones internacionales7. Representan entre el 0,1 al 3,4% de todos los tumores intracraneanos16. Los avances neuroquirúrgicos recientes hacen que la cirugía deba ser realizada antes que las otras modalidades terapéuticas36 y la radioterapia postoperatoria ha dado muy buenos resultados con sobrevidas del 70% a los 10 años20, 27. El tumor puede diseminarse por el L.C.R. y por ello se debe irradiar también todo el neuroeje4, 20. (Fig. 3)

Fig. 2: Paciente de 10 años, de sexo femenino, con ependimoma de la región pineal. A) RNM pre quirúrgica. B) RNM post quirúrgica.

Fig. 2 C. Histología
Referente al tratamiento quirúrgico somos partidarios del abordaje directo de la lesión teniendo en cuenta la baja mortalidad operatoria actual, lograda con los avances técnicos12, 17, 26, 34, 35. En 8/9 de los pacientes explorados utilizamos el abordaje transcalloso8, 12, 14 y en uno la vía suboccipital transtentorial14, 19, 22. La elección de la vía dependió de la dirección del crecimiento tumoral14.
.
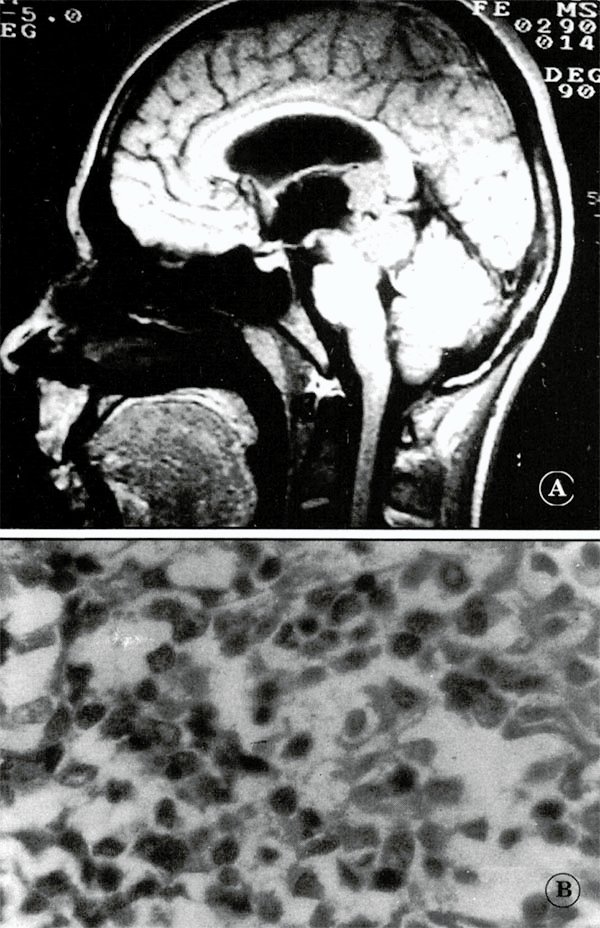
Fig. 3: Paciente de 14 años, de sexo masculino, con germinoma de la región pineal, abordado por vía suboccipital transtentorial. A) RNM. B) Histología.
No efectuamos biopsias cerebrales estereotáxicas por la dificultad que este método nos ofrece para evaluar exactamente la naturaleza histológica del tumor, teniendo en cuenta que la mayoría de ellos son mixtos y por el riesgo de hemorragia fatal durante el procedimiento6.
Concerniente a la derivación del L.C.R., en la actualidad sólo la realizamos en el postoperatorio, cuando la resección tumoral fue subtotal y persiste la obstrucción del acueducto de Silvio, porque la presencia de hidrocefalia nos facilita el abordaje a la región pineal y porque la derivación aumenta el riesgo de diseminar el tumor fuera del sistema nervioso. En nuestra serie, de los 5 pacientes derivados, 3 la requirieron en el postoperatorio por haberse resecado subtotalmente el tumor (2 pineoblastomas y 1 astrocitoma), los otros 2 enfermos fueron derivados e irradiados, con desaparición del tumor en uno y con persistencia de la neoplasia en el otro, por lo cual, este último fue llevadoa nuestra Institución para exéresis tumoral, resultando ser un teratoma benigno14, 24
BIBLIOGRAFÍA
1. Allen J: The effects of cancer therapy on the nervous system. J. Pediat 93: 903-909, 1978.
2. Arabi C, Matsumoto S: Statistical re-evaluation of pinealoma and related tumors in Japan. J Neurosurg 32: 146-149, 1969.
3. Becker LE, Hinton D: Primitive neuroectodermal tumors of the central nervous system. Hum Pathol 14: 538-550, 1983.
4. Bjornsson J, Scheithaver B W, Okasaki H, Lecch RW: Intracranial germ cell tumors; pathobiologic and inmunohistochemical aspects of 70 cases. J Neuropathol. Exp Neurol 44: 32-46, 1985.
5. Bruce DA, Schut L, Sutton LN: Pineal region tumors. In: Pediatric Neurosurgery. 2nd Edition, Eds: Mc Laurin RL, Schut L, Venes JL, Epstein FWB. Saunders Company 1989, Chapter 35, pp. 409-416.
6. Chang CG, Kageyama N, Kobayashi T, Yoshida J, Negoro M: Pineal tumors clinical diagnosis with special emphasis on the significance of pineal calcificance. Neurosurgery 8: 656-668, 1981.
7. Cohen ME, Duffner PK: International Review of Child Neurology Series. Second Edition. Rayen Press Book, 1994, chapter 16, pp. 329-346.
8. Dandy WE: An operation for the removal of pineal tumors. Surgery Gynec Obstet 33: 113-119, 1921.
9. Dearnaley DP, A'Hern RP, Whittaker S, Bloom HJG: Pineal and CNS germ cell tumors: Royal Marsden Hospital. Experience 1962-1987. Inst J Radiat Oncol Biol Phys 18: 773-788, 1990.
10.Fetell MR, Stein BM: Therapy of pineal region tumors. Neurology 1: 185-188, 1984.
11.Ganti SR, Hilal SK, Stein BM, et al: CT of pineal region tumors. AJNR 7: 97-104, 1986.
12.Guevara JA, Suárez JC, Carrea R: Tumores Intraventriculares Supratentoriales en la Infancia. Boletín de la Asoc Arg de Neurocirugía 9: 26, 1970.
13.Hirsch JF, Renier D, Czernichow P, Beneviste L, Pierre Kahn A: Medulloblastoma in childhood. Survival and Functional Results. Acta Neurochir (Wien) 48: 1-5, 1979.
14. Hoffman HJ, Yoshida M, Becker LE, Hendrick EB, Humphreys RP: Pineal region tumors in childhood. Experience at the Hospital for Sick Children. Concepts in Pediatric Neurosurgery 4: 360-386, 1983.
15.Hoffman HJ: Supratentorial brain tumors in children. In: Neurological Surgery. Ed: Youman. Second Edition. W.B. Saunders Company, 1982, vol.5, pp. 2702-2732.
16.Horowitz MB, Hall WA: Central Nervous System Germinomas. A review. Arch. Neurol 48: 652-657, 1991.
17.Horrax G: Treatment of tumors of the pineal body, experience in a series of 22 cases. Arch Neurol Psychiat, Chicago, 64: 227-242, 1950.
18.Ingraham FD, Bailey OT: Cystic Teratomas and Teratoid tumours of the central nervous system in infancy and childhood. J Neurosurg 3: 511-532, 1946.
19.Jamieson KG: Excision of pineal tumors. J Neurosurg 35: 550-553, 1971.
20.Leibel SA, Sheline GE: Tolerance of the brain and spinal cord to conventional irradiatiom. In: Gutin PH, Leibel SA, Sheline GE (eds). Radiation injury of the nervous system. Rayen Press, New York, 1991; pp. 239-256.
21.Martins AN, Johnston JS, Henry JM, Stoffel TJ, Di Chiro G: Delayed radiation necrosis of the brain. J Neurosurg 47: 336-345, 1977.
22.Poppen L: The right occipital approach to a pinealoma. J Neurosurg 25: 705-710, 1966.
23.Raimondi AJ, Tomita T: The disadvantage of prophilatic whole CNS post operative radiation therapy for medullolstoma, in multidisciplinary aspects of braIn tumor therapy. Elsevier North Holland. Biomedical Press, Amsterda, 1979, pp. 209-218.
24.Rao Yts, Medini E, Haseloun RE, Jones TK, Levitt SH: Pineal and ectopic pineal tumors: the role of radiation thrapy. Cancer 48: 708-713, 1981.
25.Rorke LB: The cerebellar medulloblastoma and its relationship to prímitive neuroectodermal tumors. J Neuropathol, Esp. Neurol 42: 1-15, 1983.
26. Rusell WO, Saches E: Pínealoms. Clínico pathologic study of seven cases with a review of the literature. Arch Path 35: 869-888, 1943.
27.Sano K, Matsutaní M: Pinealoma (germinoma) treated by direct irradiation. A Long term follop up. Child's Brain 35: 81-97, 1981.
28.Sawaya R, Hawley DK, Tobler WD, Tew JM (Jr), Chamber AA: Pineal and Third Ventricles Tumors. Neurological Surgery. Youman JR (ed.) WB Saunders Company 1990, Third Edition, vol. 5, chapter 109, pp. 3171-3203.
29.Schiffer D: Tumors and Dysontogenetic Lesions. In: Brain Tumors. Pathology and its biological correlates. Schiffer D (ed.) Springer-Verlag, 1993, chapter 21, pp. 349-404.
30.Sprung C, Baerwald R, Henkes H, Schorner W: A comparative study of CT and MRI in midline tumors of childhood and adolescence. Child's Nervous Systm 5: 102-106, 1989.
31.SuárezJC, Viano JC, Oulton CA, Zunino S: Tumores de Fosa Posterior en la Infancia. Revista Argentina
de Neurocirugía 4: 12-18, 1988.
32.SuárezJC, Sfaello ZM, Córdoba R, Viano JC, Oulton CA, Zunino S: Tumores Cerebrales en Pediatría. Revista Argentina de Neurocirugía 6: 8-14, 1991.
33.Suárez JC, Viano, JC, Herrera EJ: Cirugía de los Tumores Primarios de Tronco Cerebral. Revista Arentina de Neurocirugía 7: 26-30, 1993,
34: Susuld J, Iwabuchi T: Surgical Removal of pineal tumors (pinealomas and teratomas). Experience in a series fo 10 cases. J Neurosurg 23: 565-571, 1965.
35.Stein BM: The infratentorial supracerebellar approach to pineal lesions. J Neurosurg 35: 197-202, 1971.
36.Stein BM: Supracerebellar infratentorial approach to pineal tumors. Surg Neurol 11: 331-337, 1979.
37.Takeuchi J, Handa H, Oda Y, Uchida Y: Alphafetoprotein in íntracranial malignant teratoma. Surg Neurol 12: 400-404, 1979.
38.Vebi K, Tamaka R: Treatment and prognosis of pineal tumors; experience of 110 cases. Neurol Med Chir (Tokyo) 20: 1-26, 1980.
39.Wood JH, Zimmerman RA, Bruce DA, et al: Assessment and management of pineal region and related tumors. Surg Neurol 16: 192-195, 1981.
40.Zee C, Segall H, Apuzzo M, et al: MR imaging of pineal region neoplasms. J Comput Assist Tomogr 15: 56-61, 1991.
41. Zimmerman RA, Bílaniuk LT, Wood JH, et al: Computed Tomography of pineal, parapineal and histologically related tumors, Radiology 137: 669-677, 1980.