
Título
Autor
Fecha
Lugar de Realización
Texto
Tratamiento Quirúrgico de la Esclerosis Tuberosa en la Infancia
V. Cuccia, G. Zúccaro, F. Sosa, J. Monges
Servicio de Neurocirugía. Hospital Nacional de Pediatría "Prof. J. P. Garrahan". Buenos Aíres.
Correspondencia: Virrey Arredondo 3231 (1426) Capital separado:
SUMMARY
Between august 1987 and august 1997 we have studied 68 patients with tuberous sclerosis, 11 of which underwent surgery.
In nine patients the goal of the surgery was the exeresis of ventricular tumors.
Histologicalfindings: subependimal giant-cell astrocytoma.
In the other two patients surgery was performed for medically intractable epilepsy (frontal corticectomy and temporal lobectomy).
Its was not necessary to place shunts for hydrocephalus, although seven out of nine patients developed it. This disappeared with tumoral exeresis.
Radiological findings and surgical approaches and outcomes are discussed.
cc
Key words: Astrocytoma, Tuberous sclerosis, Hydrocephalus, Epilepsy.
Palabras clave: astrocitoma, esclerosis tuberosa, epilepsia, hidrocefalia.
INTRODUCCIÓN
La Esclerosis Tuberosa (ET) es de especial interés neuroquirúrgico por su frecuente asociación con tumores intraventriculares conocidos como astrocitomas subependimarios de células gigantes (ASCG) y también debido a que pueden provocar epilepsia médicamente intratable.
Descripta por Bourneville en 1880 y también conocida como epiloia, es una enfermedad autosómica dominante (cromosomas 9 y 16), habiéndose registrado también casos esporádicos. Su incidencia es de 1 en 10.000.
Al ser una displasia mesoectodérmica la afectación orgánica es múltiple, siendo frecuente en riñón (angiolipomas), corazón (rabdomiomas), piel y SNC, donde además de tumores cerebrales se pueden observar displasias corticales (tubers) y nódulos subependimarios.
Enmarcada clínicamente por convulsiones, retardo mental y lesiones cutáneas, representa el segundo síndrome neurocutáneo más frecuente, después de la neurofibromatosis tipo l.
MATERIAL Y MÉTODOS
Entre 1987 y 1997 se atendieron en los Servicios de Neurología y Neurocirugía del Hospital Nacional de Pediatría J. P. Garrahan 68 pacientes con ET, a 11 de los cuales se les realizó indicación quirúrgica (16%), 9 por tumores intraventriculares (13%) (8 operados y uno a la espera del turno quirúrgico) y 2 por epilepsia médicamente intratable (3%).
Fueron 7 varones y 4 mujeres, con una edad promedio de 9 años (rango 1 a 14 ).
En 8 pacientes se objetivó la triada clínica de Vogt's (convulsiones, retardo madurativo y lesiones cutáneas).
Todos los pacientes fueron estudiados con TAC cerebral y en 8 se realizó IRM.
Los hallazgos histológicos fueron: 8 ASCG y 2 displasias corticales (tubers).
El seguimiento promedio de los pacientes fue de 40 meses.
RESULTADOS
Analizamos ambos grupos de pacientes por separado:

Fig. 1. Paciente de 14 meses con epilepsia médicamente intratable por tubers corticales. Se realizó lobectomía frontal izquierda.

Fig. 2. Astrocitomas subependimarios bilaterales, Nódulo subependimario calcificado ventricular derecho.
A. Cirugía de la epilepsia.
Son 2 pacientes de sexo masculino, de 14 meses y 7 años con alteraciones en la piel, retraso madurativo y convulsiones refractarias a la medicación (20 y 60 crisis diarias respectivamente).
No presentaron hidrocefalia, ni SHE.
Ambos pacientes fueron estudiados con TAC y IRM, objetivándose en ambos tubers corticales, algunos de los cuales tenían correlato neurofisiológico como productores de crisis comiciales, lo que determinó realizar una corticectomía frontal y una lobectomía temporal estándar en cada caso. Las características radiológicas de los tubers fueron: homogéneos, sin captación del contraste y de diferentes densidades o intensidades según el tiempo de ponderación y según el caso.
El resultado quirürgico fue satisfactorio dado que se logró franca mejoría de las convulsiones en ambos casos (disminuyeron su frecuencia y su intensidad).
El hallazgo histopatológico fue: displasia cortical (tubers) y el seguimiento es de 12 y 24 meses.
B. Cirugía tumoral
Son 9 pacientes (5 varones y 4 mujeres) de 10 años de edad promedio.
El motivo de consulta se debió en 7 casos a síndrome de hipertensión endocraneana (HTE) (provocado por tumor intraventricular que generó hidrocefalia obstructiva) y en los 2 restantes, a la presencia de un tumor intraventricular con crecimiento comprobado, pero sin HTE.
Todos los pacientes tenían lesiones cutáneas, 8 presentaban convulsiones y 6 retardo mental. Todos fueron estudiados con TAC pre y postquirúrgica y a 6 se les realizó IRM. Ocho pacientes presentaron un solo tumor y un paciente presentó 2 tumores, siendo siempre de ubicación cercana al orificio de Monro (5 en ventrículo derecho, 3 en el izquierdo y un caso en el 3er. ventrículo).
El volumen tumoral promedio fue de 10 cm3.
Es interesante destacar que excepto un paciente, que llegó descompensado por hidrocefalia y debió ser operado de urgencia, todos tenían estudios radiológicos previos, con los cuales se pudo objetivar un crecimiento tumoral lento y diferenciar en el estudio inicial los nódulos, de los pequeños astrocitomas por la captación del contraste endovenoso de los tumores.
Si bien el hallazgo histológico siempre fue el mismo (ASCG), la velocidad de crecimiento tumoral fue diferente.
Las características radiológicas fueron similares para todos los tumores: TAC: 8 tumores hipo-densos y 1 isodenso, todos homogéneos y con gran realce postcontraste endovenoso. IRM: 3 tumores hiperintensos y 3 isointensos, todos homogéneos en T1 y los 3, en los cuales se agregó contraste endovenoso, se reforzaron con el mismo, 2 en forma homogénea y 1 en forma heterogénea.
Se operaron 8 de los 9 pacientes con tumores; el caso restante, con crecimiento lento demostrado y sin hidrocefalia, espera el turno quirúrgico.
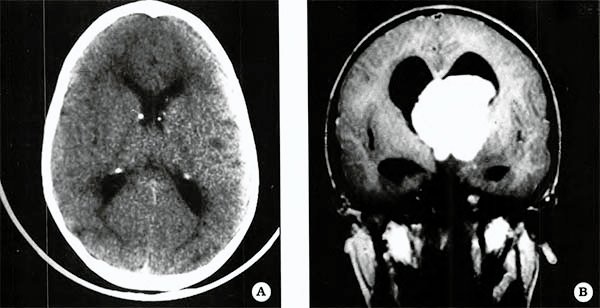

Fig. 3. En el mismo paciente: A.1992: pequeño astrocitoma ventricular izquierdo y nódulo ventricular derecho, B, 1995: importante crecimiento tumoral. C.1996: exéresis quirúrgica.
A pesar de que 7 de los 8 casos operados presentaron hidrocefalia, ninguno requirió colocación de shunt, antes ni después de la remoción tumoral. Se realizaron 5 abordajes frontales, 1 bifrontal y 2 transcallosos, lográndose 5 exéresis totales y 3 subtotales (80 a 90%).
Las complicaciones quirúrgicas fueron 3 colecciones subdurales, una hemiparesia residual y una meningitis.
No se objetivaron recidivas tumorales ni mortalidad.
DISCUSIÓN
La incidencia de tumores cerebrales en la ET2, 7,8, oscila entre el 6 y el 23%. El promedio de edad a la cirugía en nuestra serie es de 9 años, siendo infrecuente en la literatura la presentación de astrocitomas subependimarios en neonatos6.
En cuanto a los hallazgos radiológicos acordamos con Menor y col.7 que la IRM es el mejor estudio complementario para el diagnóstico y el seguimiento de estos pacientes. En los pocos pacientes que ésta resultase negativa y con sospecha clínica de ET alta, la TAC puede detectar pequeños nódulos subependimarios calcificados. Nos parece acertado incluir en los exámenes de rutina de los pacientes con ET, además de estudios neurorradiológicos seriados, exámenes oftalmológicos periódicos para prevenir trastornos visuales que se ven frecuentemente y muchas veces son irreversibles2,3.
Excepto en un paciente (que no tenía estudios previos) en todos corroboramos crecimiento tumoral y nunca observamos que un tumor se estabilice en su crecimiento como mencionan Boesel y col.1.
Diferentes son las opiniones acerca del momento de la indicación quirúrgica2,5,8,9. La nuestra es que la cirugía debe realizarse al comprobar crecimiento tumoral. Además pensamos que la hidrocefalia no debe ser tratada antes de la resección quirürgica, sin embargo otros autores consideran su corrección en forma prioritaria y a veces como único tratamiento1.
Si bien pensamos que la conducta de elección es la exéresis total del tumor, se puede considerar aceptable una exéresis subtotal amplia, que solucione la obstrucción de la circulación del LCR y que no agregue morbilidad a una patología multiorgánica y progresiva, corno lo es la ET. No observamos crecimiento importante de tumor remanente con un seguimiento promedio de 40 meses.
Si bien en nuestra serie el hallazgo histológico siempre fue el de ASCG , otros autores4 encontraron otros tipos histológicos de tumores cerebrales en esta enfermedad. Por otro lado también se describen hallazgos de ASCG en pacientes sin ET9. Consideramos este tipo de tumor (¿o displasias?) como una entidad específica de esta enfermedad y diferente de otros astrocitomas.
Por ültimo no concordamos en que los nódulos subependimarios sean los precursores tumorales5 sino que éstos son dos tipos de lesiones distintas, que pueden ser diferenciadas precozmente por la captación de contraste en la TAC2.
CONCLUSIONES
Es importante realizar TAC cerebral sin y con contraste en el momento del diagnóstico de ET, debido a que es posible diferenciar los nódulos subependimarios de los astrocitomas, aún sin mediar crecimiento; si éste se comprueba, está indicada la remoción quirúrgica tumoral antes de que se produzca hidrocefalia obstructiva, la cual en estos pacientes tiene mayor morbilidad que lo habitual (sobre todo en cuanto a deterioro visual) debido a que el diagnóstico clínico se demora por el retardo mental que gran parte de estos pacientes presenta.
Si de todas maneras se instala hidrocefalia, debe evitarse la colocación de shunt y realizar la exéresis tumoral.
Bibliografia
1. Boesel CP. Paulson GW. Kosnic EJ. Earl KM.: Brain Hamartomas and tumors associated with tuberous sclerosis. Neurosurgery 4: 410-417, 1979.
2. Di Rocco C. Iannelli A, Marchese E,: On the treatment of subependymal giant cell astrocytomas and associated hydrocephalus in tuberous sclerosis. Pediatric Neurosurg 23: 115-121, 1995.
3. Dotan SA, Trobe JD, Gebarski SS.: Visual loss in tuberous sclerosis, Neurology 41:1915-1917, 1991.
4. Frerebeau P. Benezech J. Segnarbieux F. Harbi H. Desy A. Marty-Double C.: Intraventricular tumors
in tuberous sclerosis. Child's Nerv Syst 1: 45-48, 1985.
5. Fujiwara, S. Takaki, T. Hikita, T. Nishio, S.: Subependymal giant-cell astrocytoma associated with tuberous sclerosis. Child's Nerv Syst 5: 43-44, 1989,
6. HahnJS. Bejar R. Gladson CL.: Neonatal subependymal giant cell astrocytoma associated with tuberous sclerosis: MRI, CT, and ultrasound correlation, Neurology 41: 124-128, 1991.
7. Menor E. Marti-Bonmatí L, Mulas F, Poyatos C, Cortina H.: Neuroimaging in tuberous sclerosis: a clinicoradiological evaluation in pediatric patients, Pediatric Radiology 22: 485-489, 1992.
8. Roszkowski M. Drabik K. Barszcz S, Jozwiak S.: Surgical treatment of Intraventricular tumors associated with tuberous sclerosis. Child's Nerv Syst 11: 335-339, 1995.
9. Sinson G. Sutton L, Yachnis AT, Duhalme AC, Schut L. Subependymal giant cell astrocytomas in children. Pediatric Neurosurg 20: 233-239. 1994,