
Título
Autor
Fecha
Lugar de Realización
Correspondencia
Texto
Presentación de caso
Hemangioblastomas Múltiples con una Localización en el Esplenio del Cuerpo Calloso
Jaime J. Rimoldi, Félix A. Barbone, Hugo D. Galafassi, Laura A. Bugiolacchi, Mario M. Espeche
Servicio de Neurocirugía, Hospital Rivadavia, Buenos Aires, Argentina
RESUMEN
Objetivo: presentamos un caso de hemangioblastomas múltiples con un tumor ubicado en el esplenio del cuerpo calloso. Constituye una rareza en cuanto a la localización, siendo el único reportado en la literatura neuroquirúrgica y neurorradiológica publicada en las revistas especializadas consultadas.
Descripción: paciente mujer de 18 años, portadora de hemangioblastomas múltiples que debuta su sintomatología con cuadriparesia que recupera totalmente luego de la extirpación total de dos tumores cervicales. Posteriormente se extirpó otro nódulo localizado en el rodete del cuerpo calloso, ubicación excepcional motivo de la presentación. Durante el seguimiento se observó el crecimiento de tres nódulos infratentoriales coincidentemente con su embarazo. Se realizó una tercera intervención resecando totalmente los mismos, presentando una complicación infecciosa postoperatoria que se resolvió con secuelas moderadas. Permanece actualmente libre de nuevos síntomas y sin crecimiento de otros nódulos.
Intervención: se realizaron 3 operaciones, laminectomía cervical, craneotomía occipital y craniectomía suboccipital.
Conclusión: presentamos un caso con hemangioblastomas múltiples con un tumor localizado en el esplenio del cuerpo calloso.
Se demuestra el posible crecimiento de las lesiones y su posibilidad de resección quirúrgica.
Palabras clave: hemangioblastomas, von Hippel-Lindau, cuerpo calloso, tumores supratentoriales
SUMMARY
Objective: To presenta case of multiple hemangioblastomas with a tumor located on the splenium of the corpus callosum.
Description: A 1 8-year-old female patient carrying disseminated hemangioblastomatosis suffered from tetraparesis. After the extirpation of the cervicals tumores, total recovery was observed. A second operation was performed for the extirpation of the supratentorial tumor placed in the splenial region of the corpus callosum. During the follow-up, growth of three infratentorial tumors coincidentally with pregnancy were observed. Thus, a third operation to remove them was performed. A postoperative infection developed which was solved with modera-te morbidity. Actually, the woman remains without new
symptoms and no growth of other tumors was observed.
Intervention: Three operations were performed: cervical laminectomy, occipital craniotomy and posterior fossa craniectomy.
Conclusion: We communicate an uncommon case of hemangioblastomatosis because of its location. Increase on the size of multiple lesions was demonstrated. Surgical resection was possible.
Key words: corpus callosum, hemangioblastomas, supratentorials tumors, von Hippel-Lindau syndrome.
INTRODUCCIÓN
La hemangioblastomatosis del sistema nervioso central constituye una rara enfermedad caracterizada por la presencia de tumores benignos y representa el 1,5% al 2,5%1,2 de todos los tumores endocraneales. Aproximadamente el 80% de estos tumores son únicos y ocurren esporádicamente3, el 20% se asocian con el síndrome de von HippelLindau4.
Virtualmente confinados al sistema nervioso, los sitios más comunes de localización son retina, cerebelo y médula espinal5
Las localizaciones supratentoriales son excepcionales y han sido reportados casos con hemangioblastomas localizados en nervios ópticos, región selar y lámina cuadrigeminal6,7,8
Los tumores ubicados en el tronco encefálico representan el 2% del total de los hemangioblastomas9.
Histológicamente y radiográficamente los hallazgos son similares para los de localización espinal, infra y supratentorial, clasificándose en cuatro tipos:
Tipo 1: quistes simples
Tipo 2: quiste con nódulo mural
Tipo 3: tumores sólidos
Tipo 4: tumores sólidos con pequeños quistes internos.
Se trata de tumores ricamente vascularizados y su transformación, de nodulares a quísticos, y crecimiento son posibles, como se observa en la presente comunicación, siendo éstos cambios reportados en solamente dos publicaciones previas10,11.
DESCRIPCIÓN DEL CASO
Paciente de 18 años de edad, sexo femenino sin antecedentes patológicos de importancia que ingresa a nuestro Servicio con diagnóstico de compresión medular.
Examen clínico: vigil, cuadriparesia espástica a predominio crural de rápida instauración, cefaleas frontales importantes, pares craneales conservados, sensibilidad superficial conservada y alteración bilateral de la sensibilidad profunda.
Laboratorio dentro de límites normales, no se constató poliglobulia. Examen de fondo de ojos sin presencia de alteraciones retinianas. Ecografías renal, abdominal y pélvica normales.
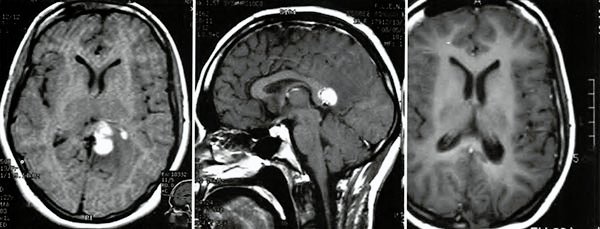
La IRM mostró dos tumoraciones en la médula cervical, con intensa vascularización en la angiografía digital; una IRM craneal mostró una turno-ración en el epitelio del cuerpo calloso (Fig 1).
Primera operación
Laminectomía C2 a C5 y apertura dural, con técnicas microneuroquirúrgica se realizó resección total de los dos tumores, cabe mencionar que la extirpación, luego de separar la aracnoides y los vasos arteriales y venosos medulares, se realizó bajo disección aguda y respetando el plano de transición tumor-tejido nervioso, fácilmente ubicable desde el comienzo de la intervención asomando a través de la pía. La intervención fue exangüe y no se produjeron complicaciones.
La paciente mejoró rápidamente su sintomatología, logrando deambular sin ayuda en aproximadamente tres meses. La IRM confirmó la extirpación total de los tumores (Fig. 2).
Anatomía patológica: numerosas células endoteliales, pericitos rodeados por membrana basal en un estroma de células claras con depósitos lipídicos, compatible con hemangioblastoma.
Segunda operación
Posición decúbito dorsal con elevación y flexión de la cabeza, craneotomía occipital unilateral izquierda, con exposición del seno sagital a fin de lograr mínima retracción occipital y abordaje por espacio interhemisférico, se llega fácilmente a la lesión que se extirpa totalmente respetando rigurosamente el plano de transición.
No se presentaron complicaciones, siendo dada de alta a la semana.
Regresó a la consulta con síndrome de hipertensión endocraneana y ataxia, sintomatología aparecida conjuntamente con su embarazo; se realizó una IRM donde se observó crecimiento de nódulos tumorales, transformación quística de uno de ellos e hidrocefalia (Fig 3).

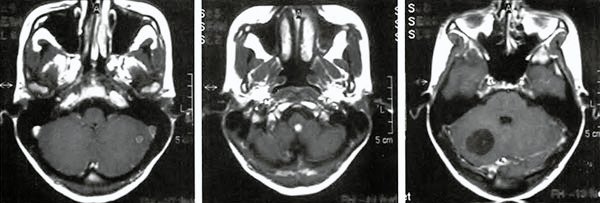
Se indicó maduración fetal y cesárea programada, tres días después se realizó resección de los dos nódulos cerebelosos y uno en tronco encefálico ubicado en el óbex.
A las 48 horas del postoperatorio la paciente se encontraba lúcida, con respiración espontánea y motilidad conservada en los cuatro miembros, desarrolla meningitis a estafilococos que se trató con ATB, según cultivos, lográndose su curación infectológica. Fue dada de alta, hallándose actualmente atáxica y en control clínico.
DISCUSIÓN
La hemangioblastomatosis múltiple constituye una rara enfermedad confinada al sistema nervioso. Aunque con mayor frecuencia los hemangio-
blastomas son únicos, su multiplicidad es posible aunque extremadamente rara. El término enfermedad de von Hippel-Lindau definía antiguamente a los pacientes con tumores multifocales asociados a por lo menos una lesión extra axial en riñón, retina, páncreas o suprarrenales, si bien hoy sabemos que las llamadas lesiones extra axiales pueden existir en 15 regiones diferentes y ser de índole histopatológica diversa desde tumores malignos de células claras, feocromocitomas y tumores de células de Langerhans, entre otras12. Actualmente la tendencia general es denominar como enfermedad de Von Hippel-Lindau a la asociación de más de un hemangioblastoma del sistema nervioso con o sin lesión visceral (extra axial) con historia familiar típica. Nuestro caso no cumple con los criterios para ser considerada como Hippel-Lindau dado que no presenta anomalías retinianas en repetidos estudios oftalmológicos, tampoco padece de localizaciones sistémicas (riñón, páncreas y glándulas suprarrenales), razón por la cual pensamos que se trata de una hemangioblastomatosis múltiple.
Efectuada la consulta genética se lo consideró como un caso esporádico.
La paciente fue evaluada mediante examen neurológico, examen clínico general, laboratorio, IRM con contraste de cerebro, órbitas, columna total, ecografías renal, abdominal y pélvica, no observándose anormalidades en otros órganos fuera del sistema nervioso.
Dadas la sintomatología, la corta edad y la inexistencia de lesiones asociadas se realizó primeramente la resección quirúrgica de los dos nódulos cervicales, que evidentemente eran responsables de los síntomas neurológicos y posteriormente el de localización supratentorial de gran tamaño y con posibilidades de complicaciones a corto plazo.
Los pequeños nódulos menores a 5 mm vistos en cerebelo y óbex se mantuvieron en observación por ser clínicamente silentes, aunque existen autores que indican tratamiento radio quirúrgico para estas lesiones pequeñas13,14; bajo las circunstancias acontecidas en la evolución de éste caso, hoy pensamos que el seguimiento semestral es lo más indicado para demostrar un crecimiento de los nódulos y extirparlos con un tamaño menor, conducta que disminuye los índices de morbimortalidad.
Sin duda que el embarazo cumplió un papel desencadenante en el crecimiento de los tumores infratentoriales.
Macroscópicamente los tumores son rojos vinosos, bien delimitados, blandos y la presencia de una fina y rica vascularización explica las hemorragias intra tumorales frecuentes con brusca expansión del tumor y la aparición rápida de sintomatología como lo observado por nosotros clínica y radiológicamente.
Microscópicamente presentan características que los hacen fácilmente distinguibles con microscopía óptica, al observarse gran cantidad de capilares de tamaño variado, separados por células tumorales sin anomalías nucleares, de aspecto espumoso por la presencia de lípidos intra citoplasmáticos; es fácilmente visible además, un claro límite del tumor con el tejido nervioso adyacente, con cambios glióticos que el neurocirujano debe saber reconocer y respetar para lograr una exéresis total y sin hemorragias15,16,17.
CONCLUSIÓN
El caso presentado es atípico y excepcional por tratarse de una localización no publicada de un hemangioblastoma.
El tratamiento del mismo consistió en la resección quirúrgica total, al igual que en otras localizaciones y pensamos que es la mejor opción frente a esta enfermedad. Si bien existen en la literatura casos tratados con radiocirugía consideramos a ésta como segunda forma de tratamiento o primeramente en aquellos de localización profunda o dentro del tronco encefálico y de pequeño volumen.
Aseguramos que el tratamiento de las lesiones sintomáticas debe ser precoz, dado que hemos demostrado su carácter evolutivo.
Es importante buscar su asociación con la enfermedad de von Hippel-Lindau, dada la característica multi-orgánica de ésta última.
El principal estudio a realizar en estos pacientes es la IRM con contraste, mediante la cual es posible observar las diferentes localizaciones, tamaño y evolución de los tumores.
Por último, si bien se trata de una enfermedad muy poco frecuente, se la debe tener en cuenta a la hora del diagnóstico diferencial de nódulos tumorales múltiples, dado que se trata de tumores benignos con posibilidades de extirpación total.
Bibliografia
1. Neumann HP, Eggert HR, Weigel K, Friedburg H, Wiestler OD, Scholimeyer P. Hemangioblastomas of the central nervous system: a 10-years study with special reference to von Hippel-Lindau syndrome. J Neurosurg 1989; 70: 24-30.
2. Fischer G, Brotchi J (eds). Intramedullary spinal cord tumors, Stuttgart. Thieme 1996; 1 -115.
3. Robert J, Weil M, Alexander O, Vortmeyer D, Zhengping Z, Svetlana D, et al. Clinical and molecular analisis of disseminated hemangioblastomatosis of the central nervous system in patients without Von Hippel Lindau diseasse: Report of four cases. J Neurosurg 2002; 96: 75-787.
4. Conway JE, Chou D, Clatterbuck RE, Bren H, Long DM, Rigamonti D. Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery 2001; 48: 55-63.
5. Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linchan WN, Zbar B. von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 1995; 194: 629-42.
6. Brodkey, JA, Buchignani JA, O' Brien TF. Hemangioblastoma of the radial nerve: case report. Neurosurgery 1995; 36: 1992-98.
7. Kerr DJ, Scheithauer BW, Miller GM, Ebersold MJ, McPhee TJ. Hemangioblastoma of the optic nerve: case report. Neurosurgery 1995; 36: 570-3.
8. Sajadi A, Tribolet De N, Unusual locations of hemangioblastomas. Case illustration. J Neurosurg 2002; 97: 727.
9. Weil RJ, Lonser RR, DeVroom HL, Wanebo JE, Oldfield EH. Surgical management of brainstem hemangioblastomas in patients with von HippelLindau disease. J Neurosurg 2003, 98: 95-105.
10. Wanebo JE, Lonser RR, Glenn GM, Oldfield E H,: The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease J Neurosurg 2003; 98: 82-94.
11. Slater A, Moore N, Huson SM. The natural history of cerebellar hemangioblastomas in von Hippel Lindau diseasse. Am J Neuroradiol 2003, 24: 1570-4.
12. Decq P, Kéravel, Y, Velasco F, (eds). Neurocirugía 1ra edición, México, JGH, 1999.
13. Chang SD, Meisel JA, Hancock SL, Martin DP, McManus M, Adler JR. Treatment of hemangioblastomas in von Hippel-Lindau disease with linear accelerator-based radiosurgery. Neurosurgery 1998; 43: 28-35.
14. Chandler HC Jr., Friedman WA. Radiosurgical treatment of a hemangioblastoma: case report. Neurosurgery 1994; 34: 353-5.
15. Malis L I. Atraumatic bloodless removal of intramedullary hemangioblastomas of the spinal cord. J Neurosurg 1988; 68: 550-3.
16. Lonser RR, Weil RJ, Wanebo JE, DeVroom HL, Oldfield EH. Surgical management of spinal cord hemangioblastomas in patients with von HippelLindau disease. J Neurosurg 2003; 98: 106-16.
17. Van Velthoven Y, Reinacher P, Klisch J, Neumann H, Glasker S. Treatment of intramedullary hemangioblastomas, with special attention to von HippelLindau disease, Neurosurgery 2003; 53: 1306-14.
COMENTARIO
Los autores comunican un caso con localizaciones múltiples de hemangioblastoma en médula cervical, esplenio del cuerpo calloso y cerebelo, sin afección oftalmológica ni visceral, ni antecedentes familiares. Los tumores fueron operados secuencialmente, de acuerdo a la sintomatología predominante, con buenos resultados.
Los hemangioblastomas son tumores poco frecuentes del sistema nervioso. En aproximadamente un 80% de los casos son lesiones únicas, esporádicas, benignas y curables con la extirpación quirúrgica. Cuando presentan localizaciones múltiples simultáneas o sucesivas, con antecedentes familiares y asociados a hemangioblastomas retinianos y tumores viscerales (carcinoma renal, feocromocitoma, tumores quísticos renales y pancreáticos, etc) corresponden a la enfermedad de von Hippel-Lindau, que es hereditaria en forma autosómica dominante y puede evolucionar en forma maligna. En los últimos años se ha identificado y localizado un gen supresor conocido como "VHL", que en la enfermedad de von Hippel-Lindau sufre mutaciones que lo inacti van, condicionando así las manifestaciones clínicas. Es probable que el caso comunicado en el presente trabajo corresponda a lo que Weil et al denominan "hemangioblastomatosis diseminada sin enfermedad de von Hippel-Lindau"1 de evolución maligna ,sin historia familiar, y con alteraciones en el gen VHL, lo que enfatizaría la necesidad de un estricto seguimiento a largo plazo de la paciente, ante la posibilidad de recidivas alejadas y/ o diseminación subaracnoidea.
Daniel H. D'Osvaldo
Instituto de Neurociencías Aplicadas´, Hospital de Clínicas "José de San Martín", Buenos Aires
1. Weil RJ, Vortmeyer AO, Zhuang Z, Pack SD, Theodore N, Erickson RK, Oldfield EH. Clinical and molecular analysis of disseminated hemangioblastomatosis of the central nervous system in patients without von Hippel-Lindau disease. Report of four cases. J Neurosurg 2002; 96: 775-87.