
Título
Autor
Fecha
Lugar de Realización
Correspondencia
Texto
Presentación de caso
Enfermedad de Von Hippel Lindau y Hemangioblastomas Espinales Asintomáticos: Descripción de un Caso, Revisión y Algoritmo
Francisco Mannará, Gustavo Baikauskas, Javier Gardella.División de Neurocirugía, Hospital Municipal General de Agudos "Juan A. Fernández", Buenos Aíres, Argentina.
Correspondencia: Av. Triunvirato 4053, 7° "B", (1431) Buenos Aires.
E-mail: fmannara@hotmail.com
RESUMEN
Objetivos: Describir un caso de enfermedad de von Hippel Lindau (VHL), realizar una revisión sobre VHL, la controversia sobre el tratamiento de los hemangioblastomas espinales asintomáticos (HEA) y diseñar un algoritmo diagnóstico terapéutico.
Descripción: Paciente de sexo masculino, 21 años de edad con antecedentes paternos de hemangioblastomas cerebeloso y cervical, que consultó por un síndrome de hipertensión endocraneana. Fue estudiado con neuroimágenes, que mostraron una lesión en fosa posterior compatible con hemangioblastoma. Se indicó cirugía.
Intervención: Se realizó una craniectomía suboccipital con resección del hemangioblastoma cerebeloso. La evolución fue favorable. En el estudio postoperatorio se encontró un HEA, que no recibió tratamiento quirúrgico. Se hizo una revisión de la literatura para actualizar la conducta ante estos casos y se elaboró un algoritmo diagnóstico terapéutico. Conclusiones: De acuerdo con el estudio y según el algoritmo elaborado, los HEA enmarcables en el diagnóstico de VHL, no deben ser inicialmente operados. Deben ser seguidos por clínica e imágenes priorizando, en su evaluación, el tamaño del quiste, su asociación con siringomielia y la tasa de crecimiento tumoral.
Palabras clave: algoritmo, enfermedad de von Hippel Lindau, facomatosis, hemangioblastomas espinales.
ABSTRACT
Objective: To describe a patient with von Hippel Lindau disease (VHL) and an asymptomatic spinal hemangioblastoma (ASH) and to make review and propose a diagnostic-therapeutic algorhytm.
Description: A 21 years old mate patient with familiar history of hemangioblastomas, that presented intracranial hypertension, with a les ion compatible with hemangioblastoma in the posterior fossa in MRI and anasymptomatic spinal hemangioblastoma (ASH),
Intervention: Through a suboccipital craniectomy the cerebellar hemangioblastoma was resected, The ASH was not operated.
Conclusion: The ASH in the course of VHL should not be initialy resected, These particular tumors must be followed with clinical examination and MRI,
Key words: Algorhytm, Phakomatosis, Spinal hemangioblastoma, von Hippel Lindau Disease
INTRODUCCIÓN
Los hemangioblastomas espinales sintomáticos son intervenidos quirúrgicamente, sin embargo, existe controversia sobre el tratamiento de los hemangioblastomas asintomáticos en los pacientes con enfermedad de von Hippel Lindaul1-7 Algunos autores opinan que sólo deben intervenirse aquellos hemangioblastomas espinales que recién comienzan a dar síntomas4, otros autores refieren que deben intervenirse quirúrgicamente los hemangioblastomas asintomáticos que presenten en el seguimiento imagenológico aumento del tamaño quístico o nodular, asociación con quiste siringomiélico, y progresión en la tasa de crecimiento tumoral5-7. Aprovechamos la descripción de un caso con enfermedad de von Hippel Lindau y hemangioblastoma espinal asintomático para revisar la literatura sobre esta entidad, analizando las diversas conductas y elaborar un algoritmo diagnóstico terapéutico.
DESCRIPCIÓN DEL CASO
Entre junio 24 y julio 8 del 2004, estuvo internado en la División de Neurocirugía de nuestro hospital un paciente de sexo masculino y 21 años de edad con antecedentes paternos de hemangioblastoma cerebeloso y cervical 3-4. Presentó en marzo del mismo año una cefalea holocraneana de gran intensidad, paroxística, acompañada de nucalgia y vómitos. Consultó y se indicó una tomo-grafía computada (TAC) de cerebro (13/6/03), que mostró una imagen hipodensa en vermis cerebeloso de 46 x 30 mm, con un nódulo hiperdenso en la pared posterior, colapso ventricular y cisternas moderadamente dilatadas.

Se realizaron imágenes por resonancia magnética (IRM) de cerebro con y sin gadolinio (19/6/ 03) (Fig 1), que mostraron un tumor con componente quístico y nódulo mural de 1.2 cm, con refuerzo postcontraste, edema, descenso amigdalino a través del orificio magno e hidrocefalia. Se realizó el diagnóstico presuntivo de hemangioblastoma cerebeloso. Al examen físico presentaba Romberg sensibilizado positivo, edema de papila bilateral y un ovillo capilar en la región temporal del ojo izquierdo (Fig 2).
Se efectuó el diagnóstico presuntivo de enfermedad de von Hippel Lindau. Se realizó una ecografía renal y de abdomen (27/6/03) para pesquisar lesiones viscerales asociadas.

Fue operado el 30/6/03, realizándose una craniectomía suboccipital con apertura de anillo occipital, exéresis del nódulo intraquístico con evacuación del quiste y plástica dural. El paciente evolucionó favorablemente y se externó el 7/7/ 03 con indicaciones médicas y un plan de estudio individual y familiar.
Se solicitó ácido vainillín mandélico en orina de 24 horas, IRM de fosa posterior y columna total con y sin gadolinio, TAC de abdomen y pelvis con y sin contraste, ecografía abdominal, agregándose p un examen oftalmológico tanto para el paciente como para la familia. Los estudios genéticos no se pudieron realizar. La TAC de abdomen mostró una pequeña imagen hipodensa en la región posterior de riñón derecho de 7 mm y otra similar de 8 mm en la región posterior de riñón izquierdo. Se observaron además, 2 pequeñas imágenes quísticas en cola pancreática, una de 6 mm y otra de 12 mm en topografía del colédoco intrapancreático. El ácido vainillín mandélico en orina de 24 horas estuvo dentro de limites normales.
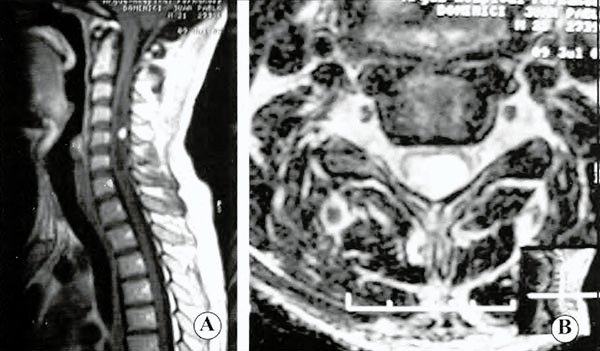
En las IRM (Fig. 3) de columna se observó una lesión intramedular cervical que se extendía desde el nivel C2-C3 hasta C6, midiendo aproximadamente 7 cm de diámetro cefalocaudal, con una estructura sólido-quística hipointensa en las imágenes ponderadas en T1, hiperintensa en las imágenes ponderadas en T2, con diagnóstico probable de hemangioblastoma medular.
En un segundo examen físico neurológico completo, con especial atención en la región metamérica afectada, realizado por un operador independiente, no se hallaron signos patológicos atribuibles a la lesión cervical. Los potenciales evocados somatosensitivos en miembros superiores fueron normales.
DISCUSIÓN
El hemangioblastoma tiene dos formas de presentación clínica: la esporádica y la familiar o enfermedad de von Hippel Lindau (VHL)1. La forma esporádica del hemangioblastoma se presenta, sobre todo, en la cuarta década de la vida con la siguiente distribución: 1-2,5% tumores intracraneales, 7-12% tumores primarios en fosa posterior, 3% en medula espinal, 2-3% en bulbo raquídeo, 1,5% supratentoriales. La enfermedad de von Hippel Lindau (VHL) es una enfermedad autosómica dominante, con 90% de penetrancia, caracterizada por la tendencia a desarrollar angiomas de retina, hemangioblastomas del sistema nervioso central (SNC), carcinoma renal de células claras (tumor de Grawitz), feocromocitomas, quistes pancreáticos, y otras lesiones tales como: tumores del saco endolinfático y quistes del epidídimo1,7.
Patología
Los hemangioblastomas son tumores benignos, bien circunscriptos, rojo cereza, sin cápsula verdadera. Frecuentemente presentan un nódulo mural y un componente quístico. Según estos hallazgos se clasifican en: sólidos o quísticos con componente mural (70 % de las lesiones cerebelosas son quísticas); los nódulos son muy vascularizados y pueden ser muy pequeños, midiendo 2 mm de diámetro. El fluido del quiste es amarillento con alto contenido proteico. Las lesiones en la unión cervicomedular y espinales generalmente son subpiales; los espinales se encuentran bien demarcados con respecto a la sustancia blanca adyacente1-2. No se han comunicado casos de malignidad histológica1.
Sato2 clasifica macroscópicamente a los hemangioblastomas en 4 grupos:
Tipo 1: corresponde al quiste simple, es generalmente cerebeloso. Lo halla en el 4,1% de los casos y plantea que el tratamiento debe ser la remoción total.
Tipo 2: asocian un quiste alargado con nódulo intramural y es el tipo más común, 60% de los casos. El interior del quiste presenta fluido xantocrómico y generalmente el nódulo se encuentra cercano a la superficie pial.
Tipo 3: hemangioblastoma sólido (26,6%), aparece como un tumor rojo esférico, que no presenta cápsula verdadera. Posee rica vascularización, cubierto por arterias y venas de drenaje que en la angiografía pueden simular una malformación arteriovenosa.
Tipo 4: es un hemangioblastoma sólido con pequeños quistes internos (9,1%). Su apariencia externa es similar al tipo 3.
Histológicamente, los nódulos' presentan 3 tipos de células:
Células endoteliales, que llenan los espacios vasculares,
Pericitos, que rodean la membrana basal,
Células estromales poligonales, que presentan citoplasma espumoso, también llamados "Zwischenzellen" o células intermedias. El origen de estas células se desconoce. Las células estromales pueden expresar citoqueratina, pero no presentan antígeno de membrana.
Se reconocen 3 tipos de hemangioblastomas según la microscopía:
Juvenil: los cuales presentan vasos dilatados y pared fina capilar;
Transicional: con pared fina capilar y vasos dilatados con células estromales sudanófilas,
Células claras: estroma vascular y células xantomatosas,
Patogenia
Hay evidencias que proponen que el hemangioblastoma es de origen disembriogénico, debido a que son encontradas en tejidos derivados del mesodermo. Según Schiffer, este tumor se debería a un defecto disembriogénico del mesénquima, que ocurriría en el tercer mes de la vida fetal y que envolvería varios órganos o tejidos del organismo, característica que comparte con otras facomatosis como ser la neurofibromatosis de von Recklinghaussen, la esclerosis tuberosa de Bournville y la angiomatosis de Sturge Weber Krabbe, A pesar de que no presenta lesiones asociadas en piel, van der Hoeve clasifica a la VHL como una facomatosis 1,2,8-11, A los hemangioblastomas localizados en cerebelo, sin otra lesión acompañante, se los denomina enfermedad de Lindau2,
Enfermedad de von Hippel-Lindau
Esta enfermedad es considerada una facomatosis, Se encuentra descripta como la enfermedad de herencia mendeliana n° 193300, cuyo gen supresor tumoral se encuentra localizado en el cromosoma 3 p 25-26 que codifica la proteína VHL polifuncional; ésta, entre otras funciones promueve el factor de crecimiento del endotelio vascular, que está incrementado en los tumores de
VHL3,5,8,10,11.
Epidemiología
La VHL presenta una incidencia de 1 cada 36,000 nacidos vivos1, aunque existe bibliografía que considera como incidencia 1 cada 93,000 habitantes.
Esta enfermedad es autosómica dominante con penetrancia casi completa a los 65 años de edad1, se manifiesta generalmente luego de la pubertad, con un pico máximo en la tercera década de la vida (más temprano que los hemangioblastomas esporádicos), con una relación 2:1 (hombre/mujer), Topográficamente, son más frecuentes los hemangioblastomas ubicados en fosa posterior; según Yasargil2 un 83% del total y preferentemente en los hemisferios cerebelosos, La incidencia en fosa posterior varía según otros autores: 72%, pero siempre como lugar preferencial1,2, El angioma o hemangioblastoma retiniano es la segunda lesión en frecuencia (59%)1,10,12,13,
La incidencia de los hemangioblastomas medulares es aproximadamente del 25%, localizándose en la región cervical con más frecuencia, siguen los torácicos y por último los hemangioblastomas lumbares5, El 96% de los hemangioblastomas espinales son posteriores, con respecto al ligamento dentado,
Los hemangioblastomas supratentoriales, antes llamados meningiomas angioblásticos presentan una incidencia menor al 1%1,2,14, También se describen hemangioblastomas en tronco encefálico, bulbo raquídeo, unión bulboespinal, y en área postrema! ,2,
Al tumor renal de células claras se lo encuentra relacionado con la enfermedad en un 28% de los casos, mientras que el feocromocitoma en un 7% de los casos,
Criterios diagnósticos de enfermedad de VHL1
Para el diagnóstico de la enfermedad se deben cumplir con los siguientes requisitos:
- uno o más hemangioblastomas dentro de SNC
- presencia de lesión visceral ( frecuentemente quiste renal y/o pancreático)
- historia familiar de la enfermedad, (incidencia familiar)
Con historia familiar de hemangioblastoma en SNC, se necesita solamente para el diagnóstico, un hemangioblastoma o una lesión visceral.
En el caso de pacientes que no presenten historia familiar de VHL, se necesitan dos o más hemangioblastomas o un hemangioblastoma y una lesión visceral.
Clínica
La clínica varía según el sitio donde se encuentre ubicada la lesión, La localización infrantentorial es la más común y frecuentemente debuta como un síndrome de hipertensión endocraneana, En varios trabajos se describe como primer síntoma la cefalea, Otros signos y síntomas son: vómitos, ataxia, alteraciones en la marcha, nistagmus, papiledema, etc,1,2,12,13,
En los hemangioblastomas de tronco se describe como síntoma la hipotensión ortostátical2, La parestesia y/o el dolor son comunes en los hemangioblastomas medulares y pueden ser seguidos de hipoestesia y paresia.
Es común que los hemangioblastomas o los angiomas de retina cursen con desprendimiento de la misma, con pérdida en la agudeza visual que pueden progresar hasta la amaurosis. Las lesiones pequeñas pueden ser asintomáticas.
El 20% de los pacientes presentan policitemia, debido a la secreción de eritropoyetina por las células intersticiales del hemangioblastoma cerebeloso, que retrógrada con la exéresis de la lesión. A pesar de la rica vascularización de la lesión, las hemorragias subaracnoideas y/o intraaxiales son infrecuentes1,2,5,12.
Laboratorio
Como están asociadas a policitemia, es imprescindible obtener un hemograma; por la relación entre la enfermedad y los feocromocitomas se debe determinar ácido vainillín mandélico en orina de 24 horas.
Diagnóstico por imágenes
TAC: se visualizan como lesiones sólidas isodensas que realzan con contraste endovenoso; los hemangioblastomas quísticos se observan como lesiones hipodensas con un nódulo mural el que realza con contraste.
IRM: es el método de elección15; muestra hemosiderina , producto del sangrado antiguo de la lesión y es ideal para evaluar fosa posterior. Las lesiones son hipointensas en T1, hiperintensas en T2, con realce con gadolinio, en particular la parte sólida y el nódulo mural.
Angiografia: denota la intensa vascularización. Es útil cuando el nódulo es demasiado pequeño para ser observado por IRM (menores a 5 mm) .Se describen cuatro patrones angiográficos1:
- nódulo vascular y quiste avascular;
- lesión vascular rodeada de quiste avascular;
- masa sólida vascular;
- nódulos vasculares múltiples.
Tratamiento
El tratamiento de elección preconizado es el quirúrgico1-3,5,6,12. La cirugía consiste en remover totalmente el nódulo mural , sin plantear la extirpación del quiste en su totalidad16. Sin embargo, debido a su naturaleza muy vascularizada , los tumores localizados en zonas vitales, o debido al sangrado profuso durante la cirugía pueden no ser extirpados en su totalidad (resección parcial)2.
Con respecto a la radiocirugía, la literatura refiere su utilidad en tumores múltiples profundos o tumores inoperables (tallo cerebral)1,2,12 y combinada con cirugía en tumores muy vascularizados, para con posterioridad a la irradiación ser removidos quirúrgicamente2.
A propósito del caso de referencia: el estudio o exploración del paciente con hemangioblastoma, enmarcable en el diagnóstico presuntivo de VHL, consistió en: ácido vainillín mandélico de 24 horas en orina, TAC con y sin contraste de abdomen y pelvis, ecografia abdominal, fondo de ojo e IRM de columna total con y sin gadolinio.
En las IRM de columna se informa una imagen intramedular cervical (C2-C3 hasta C6), con estructura sólido quística hipointensa en T1, hiperintensa en T2 con realce con gadolinio del nódulo mural en el sector posterior y central, compatible con hemangioblastoma medular.
El paciente es asintomático de la lesión espinal, por lo cual nos encontramos en la disyuntiva de "operar una imagen sin clínica, potencialmente deletérea o seguir su evolución".
Hemos ha investigado acerca de las indicaciones quirúrgicas de los hemangioblastomas, las complicaciones quirúrgicas, técnica quirúrgica, e historia natural de la enfermedad, para así poder tomar una decisión médica válida.
Los hemangioblastomas tienen períodos de crecimiento alternando con períodos quiescentes. Algunos autores postulan que la aparición de los síntomas se debe al efecto de masa producido por la lesión quística5.
Es interesante destacar que la mayoría de los hemangioblastomas presentan un componente quístico y otro sólido, y que generalmente el componente quístico aumenta de tamaño con mayor frecuencia que el componente sólido.
Otro punto importante a analizar es la tasa de crecimiento tumoral, la cual es dificil de calcular y según la localización de la lesión y si presenta síntomas o es asintomático, esta misma tasa varía3-5; por ejemplo en un estudio retrospectivo se llega a la conclusión de que la tasa de crecimiento de los hemangioblastomas sintomáticos es 1,7 veces mayor que en los asintomáticos, y que en un periodo de 32 meses el 44% de los hemangioblastomas crecerán de forma tal que podrían ser evidenciados por IRM.
Con respecto al sangrado del hemangioblastoma, los distintos trabajos refieren que tanto el sangrado subaracnoideo como el parenquimatoso medular es infrecuente, pero no presentan cifras en porcentaje.
Russel et al describen en un período de diez años una clasificación según los síntomas y signos que los pacientes presentaban en el momento del examen físico prequirúrgico; luego de la cirugía, se volvió a evaluarlos clínicamente, y clasificándolos según la evolución postquirúrgica llegaron a la conclusión de que los pacientes asintomáticos o con escaso síntomas quedaban en la misma categoría o en algunos casos disminuían en la clasificación inicial (peor evolución)4-6.
En otras comunicaciones se describen las complicaciones quirúrgicas, las cuales son: 9% fistulas de LCR, 4% infecciones, 2% meningitis aséptica, y 2% de mortalidad5.
Según la bibliografía y por lo anteriormente referido, los hemangioblastomas sintomáticos deben ser intervenidos quirúrgicamente, en el caso de los asintomáticos, en cambio, existe controversia al respecto. Van Velthoben et al en un estudio retrospectivo a 10 años, concluyen que se han de intervenir quirúrgicamente los hemangioblastomas asintomáticos asociados a pseudoquistes o los de gran tamaño, debido a que estos al seguir creciendo y convertirse en sintomáticos, no retrogradaban los síntomas al ser operados6.
Teniendo en cuenta los datos de nuestro caso y lo visto en la bibliografia decidimos realizar el siguiente algoritmo diagnóstico terapéutico Esquemas 1 y 2).
Esquema 1. Algoritmo diagnóstico-terapéutico del paciente con diagnóstico presuntivo de enfermedad de von Hippel Landau

Esquema 2. Algoritmo diagnóstico-terapéutico de pacientes con hemangioblastomas espinales

CONCLUSIÓN
1. El planteo frente a hemangioblastomas medulares en el contexto de la enfermedad de VHL, es el de intervenir quirúrgicamente a los hemangioblastomas medulares sintomáticos, y no así los asintomáticos - con excepción de asintomáticos que presenten aumento de tamaño en los controles posteriores y de aquellos asociados con pseudoquistes.
2. Los hemangioblastomas medulares asintomáticos no deben ser operados y deben ser seguidos mediante clínica y estudios por imágenes, priorizando el tamaño del quiste, la asociación con quiste siringomiélico y la tasa de crecimiento del tumor.
Bibliografía
1. Greenberg M. Tumor. En Greenberg M, editor. Handbook of Neurosurgery, 4°ed, Florida: Greenberg Graphic 1997; pp 291-3
2. Yasargil G. Vascular Tumors. En Yasargil G, editor. Microneurosurgery. New York:Thieme.1996; pp 385-9.
3. Stephane R., Phillipe D, Giraud S, Béroud C, Resche F .Central nervous system hemangioblastomas, endolimphatic sac tumor, and VHL disease. Neurosurgery 2000; 23: 1-22.
4. Russell R., Losner M, Robert J, Weil M, Wanebo J, DeVroom H et al. Surgical management of spinal cord hemangioblastomas: patients with VHL disease. J Neurosurg 2003; 98: 106-16.
5. Wanebo J, Russell R, Lonser M, Glen G, Oldfield E.The natural history of hemangioblastomas of central nervous system in patients with VHL disease. J Neurosurg 2003; 98: 82-94.
6. Van Belthoven V, Reinacher P, Klisch J, Neumann H, Glásker S. Treatment of intramedullary hemangioblastomas, with special attention to von Hippel Lindau disease. Neurosurgery 2003; 53:1306-14.
7. Pietilo T, Stendel R., Schilling H, Krznauc I, Brock M. Surgical treatment of spinal hemangioblastoma. Acta Neurochir (Wien) 2000;142: 879-86.
8. Goebel H. Molecular oncologic and therapeutic spectrum of VHL disease. Neurosurg Rey 2000; 23: 23-4
9. Couch V, Lindor N, Karner P, Michels V. von Hippel Lindau disease. Mayo Clinic Proc 2000; 75: 265-72.
10. Seizinger BR., Smith DI, Filling-Katz MR, Neumann H, Green J, Choyke PL et al. Genetic markers refine diagnostic criteria and provide insights into the genetics of VHL disease. Procl Natl Acad Sci USA 1991; 88: 2864-8.
11. Gutjohr E. von Hippel Lindau disease. Neurosurg Rey 2000; 23: 25-9.
12. Alfaro Giner A. Anomalias del desarrollo del SNC. En Farreras V, Rozman C, editors .Medicina Interna ,13° ed, Madrid. 1997, p 1480.
13. Berger M, Kross J. Tumor. En Youmans J, editor. Neurological Surgery, 4a ed. California: Saunders Company, 1996, pp 2703-8.
14. Wasenko J, Rodziewicz G. Suprasellar hemangioblastoma in VHL disease a case report. Science 2003; 27: 18-22.
15. Bao Cheng C, Satoshi T, Kasutoshi H, Matakazu F. MR findings in spinal hemangioblastoma: correlation with symptoms and with angiographic and surgical findings, 2001; 22: 206-7,
16. Malis L, Atraumatic bloodless removal of intramedullary hemangioblastomas of the spinal cord, J, Neurosurg (Spine) 2002; 97: 1-6.
17. Huson S, Harper P, Hourihon M, Cole G, Weeks RD, Compstom DA, Cerebellar hemangioblastomas and VHL disease, Brain 1986; 109:1297-310,
18. Hermies M, Cotton F, Saint Pierre G, Jouvet A, Ongolo-Zolo P, Fischer G et al, Myelopathy and sciatica induced by an extradural S1 root hemangioblastoma , Neurorradiology 2002; 44: 494-8.
19. Grunberg A, Rodesch G, Hunt M, Carlier R,, Doyon D, Magnetic resonance imaging of intraspinal hemangioblastoma, Neurochirurgie 1994; 40: 15564,
20, Pluto R, Iuliano B, DeVroom H, Nguyen T, Oldfield
E. Comparison of anterior and posterior surgical approaches in treatment of ventral spinal hemangioblastomas in patients with VHL disease, J Neurosurg 2003; 98: 117-24,
21, Reyns N, Assaker R,, Louis E, Lejeune JP, Leptomeningeal hemangioblastomatosis in a case of VHL disease: case report, J Neurosurg 2003; 52: 1212-5,
COMENTARIO
Sobre la base de un caso de un paciente de 21 años, con antecedentes paternos de Hemangioblastoma Cerebeloso y Cervical, los autores han realizado un detallado análisis del caso clínico y una prolija revisión bibliográfica de esta entidad, poco frecuente dentro de la patología intracraneana, destacando sus dos formas de presentación clínica, la esporádica y la familiar o enfermedad de von Hippel Lindau. Su análisis patológico, patogenia, criterios diagnósticos de cada una de ellas, y las alternativas de estudio y tratamiento de la formas intracraneanas y espinales es pormenorizado, proponiendo un algoritmo diagnóstico y terapeútico para cada una de ellas, Personalmente pienso que los algoritmos deben ser reservados para aquellas entidades de mayor incidencia estadística y con diferentes variables de estudio y alternativas terapéuticas que justifiquen intentos de normatización, y de cuyo análisis y resultados pueda llegarse a conclusiones que faciliten el quehacer neuroquirúrgico, Dejando el análisis personalizado para las entidades de baja incidencia, donde los intentos de dogmatizarlas pueden quedar descolocados, correspondiendo en ellas el análisis particular de cada caso, Esta opinión no va en desmedro del trabajo de los autores, que presentan una excelente puesta al día de esta entidad , destacándose la bibliografía utilizada, que seguramente servirá de referencia cuando estemos frente a casos de este tipo de facomatosis.
Prof. Dr, Jorge D. Oviedo
Hospital Alemán Buenos Aires,