
Título
Autor
Fecha
Lugar de Realización
Correspondencia
Texto
REV ARGENT NEUROC. VOL. 33, N° 3: 52-55 | 2019
REPORTE DE CASO
Neurocitoma central: a propósito de un caso
Wellerson Sabat Rodrigues, Matias Baldoncini, Santiago Giusta, Maria V. Montero,
Maximiliano Zarco, Luciana Perren, Agustin Conde
Servicio de Neurocirugía Hospital Petrona V. de Cordero de San Fernando. Buenos Aires, Argentina.
RESUMEN
Introducción: El neurocitoma central fue descripto por primera vez en 1982 por Hassoun et al. Se trata de una neoplasia rara, bien diferenciada del sistema nervioso central de origen neuroectodermico, ubicado más comúnmente a nivel del sistema ventricular, típicamente adyacente al foramen de Monro. Cursa generalmente con síntomas de hipertensión intracraneal secundaria a hidrocefalia no comunicante. Afecta generalmente a adultos jóvenes, con edad de presentación media de 29 años en las mayores series descriptas.
Objetivos: Describir y presentar un caso de tumor cerebral específico, cuya importancia se da debido a su baja prevalencia y escasa casuística relatada en la literatura.
Descripción del caso: En el presente artículo describimos un caso de una paciente de 35 años diagnosticada incidentalmente con una lesión ocupante de espacio a nivel del ventrículo lateral izquierdo redondeada, heterogénea, de bordes netos con dimensiones de 40x30x30 mm. La paciente fue intervenida quirúrgicamente para su resección. Se realizó abordaje interhemisférico transcalloso homolateral. Sin intercurrencias post-quirúrgicas fue dada de alta 4 días luego de la cirugía. El informe anatomo-patológico demostró tratarse de un Neurocitoma Central. Se comparó nuestro caso con lo descripto en la literatura.
Conclusión: El neurocitoma central a pesar de no ser una patología prevalente, debe ser conocido en profundidad por los neurocirujanos, ya que su correcto manejo afecta directamente al pronóstico de los pacientes.
Palabras claves: Neurocitoma Central; Abordaje Transcalloso; Microcirugía
ABSTRACT
Introduction: The central neurocytoma was first described in 1982 by Hassoun et al. It is a rare, well-differentiated neoplasm of the central nervous system of neuroectodermal origin, located most commonly at the level of the ventricular system, typically adjacent to the foramen of Monro. It usually presents with symptoms of intracranial hypertension secondary to non-communicating hydrocephalus. It generally affects young adults, with an average age of presentation of 29 years in the largest series described.
Objetives: Describe and present one case of specific brain tumor, which is important due to its your low prevalence and scarce casuistic in the literature.
Case presentation: In the present article, we describe a case of a female 35-year-old patient diagnosed incidentally with a heterogeneus rounded space-occupying lesion at the level of the left lateral ventricle, with net edges and dimensions of 40x30x30mm. The patient was surgically intervened for tumoral resection. We opteded to use a homolateral transcallosal interhemisferic approach. Without post-surgical complications, she was discharged 4 days after surgery. The anatomo-pathological report proved to be a Central Neurocytoma. We compared our case with the existing publications.
Conclusion: Despite being an uncommon tumor, Central Neurocytoma must be well understood by every neurosurgeon, considering that its adequated management influences the patient´s prognosis directly.
Key words: Central Neurocytoma; Transcallosal Approach; Microsurgery
Wellerson Sabat Rodrigues
wellerson.med@hotmail.com
Recibido: Mayo de 2019.
Aceptado: Julio de 2019.
INTRODUCCIÓN
El neurocitoma central fue descripto por primera vez en 1982 por Hassoun et al.4. Se trata de una neoplasia rara, bien diferenciada del sistema nervioso central de origen neuroectodermico5. Corresponden a aproximadamente 0,25% - 0,5% de todos los tumores intracraneanos8,13. No presentan predilección por sexo13 y afectan más comúnmente a adultos jóvenes con edad media de presentación alrededor de los 29 años2 (70% de los casos entre 20 y 40 años). Entre las formas más comunes de presentación se encuentran los signos y síntomas relacionados al aumento de la presión endocraneana secundaria a hidrocefalia obstructiva, también puede presentarse con trastornos visuales o mentales5 o aún, menos comúnmente, como lesión asociada a hemorragia intraventricular espontanea3. En el estudio de resonancia magnética se puede apreciar típicamente una lesión expansiva a nivel del sistema ventricular, generalmente en los ventrículos laterales en sitios adyacentes al foramen de Monro (50%)7, dicha lesión suele tener un aspecto heterogéneo, isointensa en T1 con relación a la sustancia gris cerebral, con variable captación de contraste de forma heterogénea, en T2 y en el FLAIR se presenta con alta señal y generalmente con múltiples áreas quísticas (aspecto de burbujas) que son atenuadas casi en su totalidad en FLAIR.
El tratamiento se basa en la resección de la lesión; según refiere Schild et al.14, la sobrevida a los 5 años fue del 90% con resecciones totales, mientras que en resecciones subtotales fue del 77%.
El diagnóstico anatomo-patológico se basa en la inmunohistoquímica de los antígenos neuronales como la sinaptofisina y la enolasa específica de neuronas 2 (NSE 2)6. Las características de dichas lesiones en la microscopia óptica suelen ser muy similares a los oligodendrogliomas, pudiendo confundirse fácilmente dichas entidades si no se realizan técnicas de inmunomarcación6,8.
PRESENTACIÓN DEL CASO
Paciente de sexo femenino de 35 años de edad quien debido a accidente automovilístico efectuó estudio tomográfico de encéfalo donde se observó lesión intraventricular redondeada, de gran tamaño, heterogénea, predominantemente isodensa con focos hipodensos en su interior. Fue derivada a servicio de Neurocirugía, se realizó estudio de resonancia magnética del encéfalo con y sin contraste.
Al interrogatorio paciente sin antecedentes patológicos de relevancia, asintomática, a la exploración física: Lúcida, eupsíquica, sin trastornos motores o sensitivos, pares craneales sin alteraciones.
En el estudio de resonancia magnética del encéfalo se observó lesión redondeada, intraventricular a nivel de ventrículo lateral izquierdo, de bordes netos, de aproximadamente 40x30x30 mm, de señal heterogénea, predominantemente isointensa respecto a sustancia gris en T1 con presencia de focos de baja señal en su interior. En T2 se observa lesión predominantemente hiperintensa, múltiples imágenes de aspecto quístico en su interior (aspecto de burbujas) que en el FLAIR dichas imágenes quísticas son atenuadas. Tras la administración de contraste endovenoso se observa moderada y heterogénea captación del mismo (fig. 1).
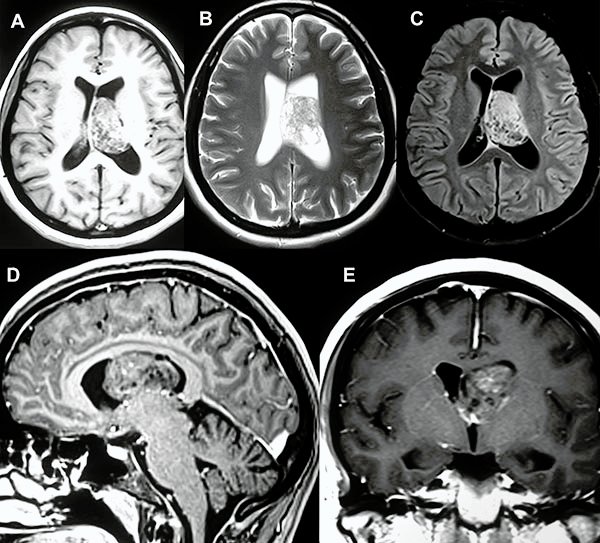
Figura 1: A, B y C) Cortes axiales en las secuencias ponderadas en T1, T2 y FLAIR respectivamente. D) Corte sagital de secuencia ponderada en T1 con contraste. E) Corte coronal de secuencia ponderada en T1 con contraste.
Se decide la conducta quirúrgica, se opta por el abordaje interhemisférico transcalloso homolateral1 ya que la lesión desplazaba el septum pellucidum y se sospechaba adherencia en el mismo.
Se realizó callosotomía anterior e inmediatamente por debajo del epéndimo del techo del ventrículo lateral se observó lesión de coloración grisácea oscura, friable y con abundante vascularización. Tras el debulking intratumoral, se logra reducir su volumen, separándolo de las estructuras intraventriculares circundantes. Con la ayuda de microespéculos se inspecciona la asta anterior, cuerpo y atrio de ventrículo lateral izquierdo sin evidencia de lesión residual (fig. 2).

Figura 2: A) Posicionamiento quirúrgico. B) Aspecto de la craneotomía del abordaje interhemisférico, previo a la apertura dural. C) Corredor interhemisférico homolateral con visualización del cuerpo calloso. D) Callosotomía y visualización de lesión de coloración oscura subyacente. E) Aspecto microquirúrgico post-exéresis de dicha lesión. F) Con la utilización de espéculos espejados se busca la presencia de restos tumorales, no hallándose lesión macroscópica residual.
Paciente permaneció internada en terapia intensiva por 48h, pasa a sala general por 48h más, sin intercurrencias y sin déficits neurológicos (fig. 3), es dada de alta hospitalaria 96h después del procedimiento quirúrgico.

Figura 3: Paciente en el cuarto día post quirúrgico, sin foco neurológico. Foto tomada con el consentimiento de la paciente, momentos antes de ser externada.
En la RM de encéfalo control, realizada 20 días luego de la cirugía, se observa huella de callosotomía anterior sin lesión focal intraventricular residual aparente (fig. 4).

Figura 4: A, B y C) Cortes axial, sagital y coronal pre quirúrgicos ponderados en T1 con contraste. D, E y F) Cortes axial, sagital y coronal post quirúrgicos ponderados en T1 con contraste.
El estudio anatomo-patológico mostró una proliferación neoplásica constituida por elementos de núcleo esférico y citoplasma claro con una disposición difusa, compacta y de moderada densidad celular, sin necrosis, anaplasia o incremento del número mitótico (fig. 5). Se pudo observar un límite nítido con el parénquima celular. Fue realizada inmunomarcación para los siguientes elementos: Sinaptofisina (SYN), Antígeno nuclear neuronal (Neu N), Proteína fibrilar ácida glial (GFAP) y Ki-67, se observó el siguiente patrón de marcación.
SYN: +++Neu N: +++GFAP: ±nnKi-67: 6%
Se confirmó, con dichos elementos, la hipótesis diagnóstica de Neurocitoma central.
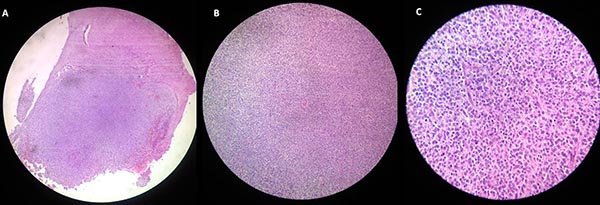
Figura 5: A) Cortes histológicos con aumento de 4x de lesión teñida con hematoxilina y eosina. B) Aumento de 10x. C) Aumento de 40x.
DISCUSIÓN
Nuestro caso coincide, tanto del punto de vista radiológico como anatomo-patológico, con lo relatado en la literatura sobre neurocitoma central. En nuestro caso el tumor se hallaba en el ventrículo lateral izquierdo, adyacente al foramen de Monro, siendo que dicha localización fue la más comúnmente observada7.
En la resonancia magnética el tumor presentaba aspecto solido con áreas quísticas y de bordes bien delimitados, la captación de contraste era moderada y heterogénea como descripto comúnmente en la literatura9,15, los aspectos radiológicos de dichos tumores fueron revisados y comparados9.
Del punto de vista histopatológico, fue imprescindible la utilización de técnicas de inmunohistoquímica para la correcta tipificación del tumor ya que las características del mismo eran fácilmente confundibles con otras entidades, no pudiéndose llegar a un diagnóstico de certeza sin la utilización de dichas técnicas, como es mencionado en la literatura6,8.
En inmunohistoquímica, la sinaptofisina es considerada el principal marcador presente en los neurocitomas, donde su intensa positividad en nuestro caso ayudó a comprobar el diagnóstico de certeza para neurocitoma central.
Un punto clave a destacarse en nuestro caso es el elevado índice de proliferación celular, marcada por el Ki-67 (6%); en la literatura se encuentra descripto una relación de peor pronóstico en los casos que presentan un índice de proliferación celular elevada comparado con los que no lo presentan10. El seguimiento a largo plazo de nuestra paciente nos dará la posibilidad de comparar si dicha relación se aplicará.
Desde el punto de vista clínico, se encontró una discrepancia en la forma de presentación ya que en nuestro caso el diagnóstico fue producto de un evento incidental y no secundario a clínica neurológica; por dicha razón no fue posible comparar los síntomas referidos en la literatura con la presentación de nuestro caso por lo antes expuesto.
La resección quirúrgica completa parece ser la mejor opción de tratamiento para el neurocitoma central. En los casos de resección incompleta, la radioterapia puede beneficiar los pacientes11. Si bien el neurocitoma central es considerado un tumor benigno, cuando se observa un índice de proliferación elevado (>2%) dicha entidad podría presentar un comportamiento agresivo10.
CONCLUSIÓN
Si bien el neurocitoma central es una entidad poco común, es fundamental que el neurocirujano conozca en profundidad dicha patología ya que su correcto diagnóstico y tratamiento son elementos clave para el mejor pronóstico y sobrevida de los pacientes. No obstante el diagnóstico no siempre se puede realizar fácilmente. Es importante un seguimiento a largo plazo de los pacientes debido a que el comportamiento de dichos tumores aún no es profundamente conocido, principalmente en aquellos que presentan un índice elevado de proliferación celular, donde su comportamiento puede ser incierto y agresivo.
Al ser lesiones que presentan una gran vascularización y localización profunda dentro de los ventrículos, constituyen desafíos neuroquirúrgicos para su remoción completa. Vale finalmente destacar que si bien hemos utilizado espéculos angulados para corroborar la resección completa, también se puede realizar esto con instrumental endoscópico angulado.
BIBLIOGRAFÍA
- Baldoncini Matias et al. Anatomia microquirúrgica y abordajes al central core cerebral. REV ARGENT NEUROC 2019 VOL. 33, N° 1: 1-13.
- Figarella-Branger D, Söylemezoglu F, Kleihues P, Hassoun J. Cantral neurocytoma. In Kleihues P, Cavenee WK. (ed). Pathology and genetics of tumours of the nervous system. Lyon: IARCPress, 2000:107-109.
- Hanel, Ricardo Alexandre et al. Neurocitoma central com apresentação incomum por hemorragia intraventricular: relato de caso. Arq. Neuro-Psiquiatr. [online]. 2001, vol.59, n.3ª.
- Hassoun J, Gambarelli D, Grisoli F, Pellet W, Salamon G, Pelliser JF, Toga M. Central neurocytoma. An electron-microscopic study of two cases. Acta Neuropathol 1982;56:151-156.
- Hassoun J, Söylemezoglu F, Gambarelli D, Figarella Branger D, von Ammon K, Kleihues P. Central neurocytoma: a synopsis of clinical and histological features. Brain Patholology 1993;3:297-306.
- Katati MJ, Vílchez R, Ros B, Horcajadas A, Arráez MA, Arjona V. Central neurocytoma: analysis of three cases and review of the literature. Rev Neurol 1999;28:713-717.
- Koeller KK, Sandberg GD. From the archives of the AFIP. Cerebral intraventricular neoplasms: radiologic-pathologic correlation. Radiographics. 22 (6): 1473-505.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK "WHO Classification of Tumours of the Central Nervous System. 4th Edition Revised" ISBN: 9789283244929.
- Mackenzie IR. Central Neurocytoma: histologic atypia, proliferation potential, and clinical outcome. Cancer. 1999 Apr 1;85(7):1606-10.
- Nishio S, Morioka T, Suzuki S, Fukui M. Tumours around the foramen of Monro: clinical and neuroimaging features and their differential diagnosis. J Clin Neurosci 2002; 9:137-141.
- Rades D, Fehlauer F. Treatment options for central neurocytoma. Neurology 2002;59:1268-1270.
- Robbins P, SegalA, Narula S, et al. Central neurocytoma: a clinicopathological, immunohistochemical and ultrastructural study of 7 cases. Pathol Res Pract 1995;191:100-111.
- Smith A, Smirniotopoulos J, Horkanyne-Szakaly I. From the Radiologic Pathology Archives: Intraventricular Neoplasms: Radiologic-Pathologic Correlation. Radiographics. 2013;33 (1): 21-43.
- Schild SE, Scheithauer BW, Haddock MG, Schiff D, Burger PC, Wong WW, Lyons MK. Central neurocytomas. Cancer 1997;79:790-795.
- Zhang B, Luo B, Zhang Z, Sun G, Wen J. Central neurocytoma: a clinicopathological and neuroradiological study.Neuroradiology 2004;46:888-895.
COMENTARIO
Los autores comunican una paciente portadora de un tumor muy poco frecuente como es el neurocitoma tratada exitosamente mediante cirugía resectiva.
El neurocitoma es una neoplasia de origen neuroepitelial, cuya localización preferencial es el sistema ventricular adyacente al foramen de Monro y al septum pellucidum; mientras que aproximadamente 10% de los neurocitomas pueden ser extraventriculares y asentar en cualquiera de los lóbulos cerebrales, región selar,o en el cerebelo.
El Gold standard de tratamiento en cualquier caso es la resección completa quirúrgica sea a cielo abierto como se realizó en este caso, o vía endoscópica, lográndose sobrevidas del 99% en 5 años de follow-up. La radiocirugía se reserva como alternativa para remanencias o recurrencias. Dado lo infrecuente de este tipo histopatológico, se recomienda seguimiento y control a largo plazo.
Alejandra T. Rabadán
Instituto de Investigaciones Médicas A. Lanari. C.A.B.A., Argentina.
COMENTARIO
El autor describe un caso de neurocitoma central, diagnosticado casualmente en contexto de un accidente de tránsito, que por medio de un abordaje interhemisférico-transcalloso, logra una resección total, con excelente evolución clínica. Este tipo infrecuente de tumor es categorizado por la OMS como grado II, comprende el 0,1-0,5% de todos los tumores cerebrales y afecta principalmente a adultos jóvenes en la tercera década de la vida.
Tres puntos importantes surgen de la lectura del artículo: el diagnóstico de certeza requiere técnicas inmunohistoquímicas, ya que la microscopia tiene dificultades para diferenciarlo de los ependimomas y los oligodendrogliomas. Son Sinaptofisina positiva y Enolasa neurona específica, Vimentina y Antígeno de membrana epitelial (de su sigla en inglés “EMA”) negativas. En segundo lugar, es de notable utilidad conocer el índice de proliferación celular para prever comportamientos más agresivos y riesgo aumentado de recidiva. Un ki-67 menor del 2% tiene una sobrevida a los 10 años del 90% mientras que en los casos con ki-67 mayor al 2%, la sobrevida desciende al 63%. En tercer lugar y como refiere el autor, este tipo de lesión pueden ser abordadas por el abordaje descripto, a través de una resección endoscópica pura (en tumores más pequeños) o a través de abordajes transcallosos con apoyo endoscópico. Si bien está descripto el uso de terapias actínicas complementarias en los casos con resecciones subtotales y recidivantes, la cirugía sigue teniendo un papel protagónico ante este tipo de casos.
Tomás Funes
Sanatorio Anchorena. C.A.B.A., Argentina.
BIBLIOGRAFÍA
- Pan DH, Lee CC. “The management of incidental central neurocitoma”. Neurosurg Clin N Am. 2015 Jan; 26(1): 57-66.
- Seung JL, Timothy T, Cheng H, Carlito L, Lawrance KC, Sabrin S, David JS, William H, Todd LS, Minsu K, Isaac Y. “Central Neurocytoma: A Review of Clinical Management and Histopathologic Features”. Brain Tumor Res Treat. 2016 Oct; 4(2): 49-57.
COMENTARIO
Aunque el pronóstico general de los neurocitomas es bastante favorable; en la clasificación de la OMS 2007 y 2016 son tumores de G II y por lo tanto pueden recidivar o progresar a los 10 años, independientemente del margen de resección quirúrgica.
La denominación neurocitoma atípico no existe como tal en ninguna de las dos clasificaciones, la misma surge del probable valor pronóstico del MIB-1/KI-67 cuando es > 2%, ya que podría ser un indicador consistente de progresión o recidiva tumoral, y por lo tanto un indicador de co adyuvancia con Rdt aún en tumores con margen de resección sano. Así la resección total (GTR) es un indicador de curación inconsistente con un MIB-1 > 2%. Los protocolos actuales indican que a Rdt mejora el control local de la enfermedad, pero no la supervivencia.
Martin A. Saez
Lomas de Zamora, Buenos Aires, Argentina.